

















The emergence of regenerative medicine as a viable therapy class has created an urgency to review and improve upon the current quality systems distribution standards that are employed in support of these therapies.
Industry efforts are already underway through both the Alliance for Regenerative Medicine (“ARM”) and the Foundation for the Accreditation for Cellular Therapy (“FACT”) whom have instituted review bodies to analyze all aspects of the collection, manufacture, transportation, and administration of regenerative medicines. Moreover, emerging Chain of ComplianceTM requirements for regenerative medicine distribution are rapidly becoming the standard for ensuring product integrity. This article will review the current and future required quality systems standards for the industry.
Regenerative Medicine Advanced Therapy (RMAT) designation is a major emerging opportunity, in many cases accelerating clinical progression for personalized therapies for diseases from hemophilia to cancer to as little as four years. This acceleration is driving companies and organizations to rapidly employ quality systems and infrastructure supporting clinical and commercial distribution in time frames that put significant stresses on an organization. Effective implementation of current and future regulatory considerations will be critical in the successful launch and management of the risks in distributing these therapies. The quality systems supporting these therapies must adapt and mature in order to effectively manage this risk. A review of the key systems requirements are as follows.

The quality systems requirements and key guiding regulations can be broken down into the following key structural components:
- Quality Systems Architecture: ISO 9001 2015, ICH Q10, and WHO Annex 5 all contribute to the key components required for a comprehensive quality systems approach that can be effectively utilized in establishing the architecture for a pharmaceutical quality system appropriate for the distribution of regenerative therapies. While the standards listed were established for the development and manufacture of pharmaceutical drug substance and drug product, the same basic standards apply to supply chain systems, equipment, and logistics management. Additional benefits are the fact that Q10 embraces a risk-based approach to quality, which marries well with Quality by Design (“QbD”) elements related to risk management via equipment performance as well as in field logistics.
- Quality Systems Design: ICH Q8-10 are complementary to one another and provide a foundation for the design of a quality system that is based on a QbD and Process Analytical Technology (“PAT”) methodology1. This approach, which allows one to address multi-dimensional external impacts on product integrity is well suited for managing risk related to complex logistics and in field risk management.
- QbD, Risk Management: Automating risk management through effective use of QbD using real-time informatics is a critical consideration in support of a commercial product in the field of regenerative medicine. Informatics to capture, interpret and predict, as well as to provide automated alerts and pre-defined escalation protocols based on the nature of the data captured for hundreds if not thousands of concurrent shipments will be critical in the effective launch of these therapies. QbD processes provide the foundation enabling such an approach to be effective.2.
- Document Control: 21 CFR Part 11, 21 CFR 1271, ICH Q7, and WHO Annex 5 provide an effective framework for the establishment of a records management system as well as the controls associated with data collection, management, as well as ensuring that companies and organizations implement good business practices by defining the criteria under which electronic records are accurate, authentic, and non-alterable.
- Electronic Equipment and Software Calibration/Validation: In addition to effective records management, ICH Q7 also dictates the calibration (5.3) and validation (5.4) of the equipment and software utilized in collection of temperature, location, and equipment traceability key to Chain of ComplianceTM and QbD processes3.

- Equipment Validation & Traceability: Regenerative medicine requires end-to-end traceability: everything from Chain of Custody, to Chain of Condition, to Chain of Identity as well as Chain of ComplianceTM, which is the traceability of the equipment, processes, and logistics handling used in managing the environmental control of the therapy while it is in transit4. 21 CFR 210, 21 CFR 211, EMA <2013/C 343/10>, and ICH Q7 provide the core components that need to be considered in the establishment of a Chain of ComplianceTM competency in support of regenerative medicine distribution. Specifically, section 211.67 equipment cleaning and maintenance should be considered.
- Facilities & Personnel: Relevant Good Manufacturing Practice (“GMP”) standards in support of clinical and commercial product distribution related to personnel and facility management, equipment cleaning, storage and maintenance, as well as supplier and materials management are covered under 21 CFR 210, 21 CFR 211, EMA <2013/C 343/10>, as well as USP <1079>. Key anticipated emerging requirements related to equipment maintenance and traceability in support of Chain of ComplianceTM standards are covered specifically in 21 CFR 210 and 211.
- Distribution & Logistics: USP <1079> is a key minimum consideration in the distribution and storage of drug product supported additionally via EMA <2013/C 343/10>, WHO Annex 9, as well as ISTA 3A,7D, 7E. These regulations in aggregate support the basic temperature and logistics management requirements including, but not limited to equipment performance, temperature management as well as lane qualification and validation. While necessary, these considerations should be augmented with a QbD methodology to manage in field risk due to 3rd party handling and at a minimum need to include active temperature, equipment, and location monitoring.
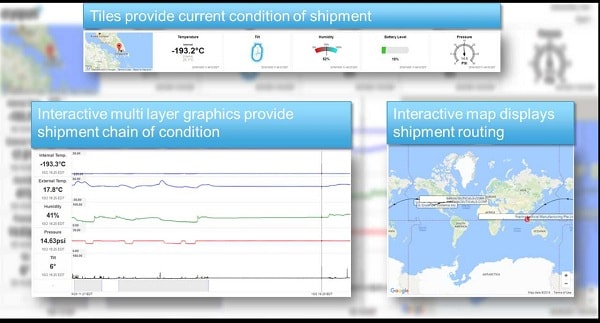
- Contamination & Packaging: Another newly emerging consideration related to cross contamination specific to cell and gene therapies is discussed in 21 CFR 600, and additionally supported by EMA <2013/C 343/10>, 21 CFR 211.67, and 21 CFR 1271. Consensus is emerging that all equipment utilized in the distribution of blood and cell materials and final product must be transported in equipment verified as only ever transporting human clinical and commercial product in order to minimize contamination risk due to non-human commodities.
The ability to effectively establish and support a comprehensive quality system in conjunction with regenerative medicine programs will significantly improve one’s logistics and distribution risk management capabilities. Additionally, effective use of QbD and Chain of ComplianceTM will provide a platform for proactive quality management processes to be employed and will minimize the risk of having to upgrade one’s systems in support of future anticipated regulatory requirements.
Moreover, complete implementation of a quality system addressing the aforementioned structural components not only will effectively manage the risk of product failure, having complete systems and data capture gives you the ability to learn and improve over time to improve your systems and processes.
1http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf
2 https://www.worldpharmatoday.com/Articles/the-arrival-of-quality-by-design-in-logistics
3 https://www.fda.gov/downloads/Drugs/…/Guidances/ucm073497.pdf
4 http://www.contractpharma.com/contents/view_features/2018-06-12/the-chain-of-compliance/



{kind=link}