

















The numbers are daunting – 15% of the global population suffers from hearing loss, including a significant number of children, teenagers, and the elderly. Hearing loss presents a growing public health problem, due to ageing and an urban way of life that features excessive noise exposure.
In addition to reducing quality-of-life and employment prospects, hearing loss poses a safety concern for those afflicted and is strongly linked to cognitive decline in the elderly. Hearing loss, which is often associated with tinnitus, may result from genetic causes, complications at birth, certain infectious diseases, chronic ear infections, use of particular drugs, exposure to excessive noise, and ageing. Hearing loss can also be induced by several classes of therapeutic compounds that are toxic to the cellular apparatus of hearing, an adverse side effect known as ototoxicity. Notably, they include aminoglycoside antibiotics (e.g. gentamycin) and certain chemotherapeutic agents (e.g. cisplatin). Development of novel drugs should include auditory safety assessments, while greater efforts are needed to prevent some of the serious auditory side effects caused by existing treatments.
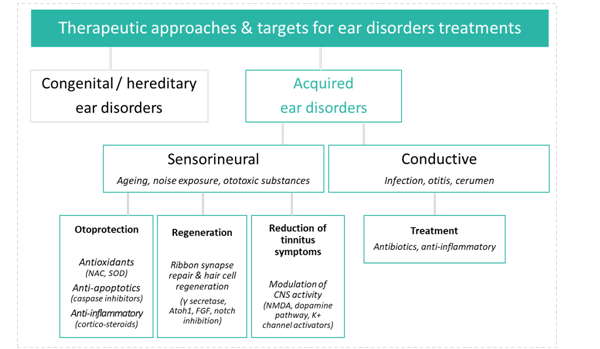
Hearing research and development aims to prevent hearing loss, halt its progression, or reverse deficits through a range of approaches. Unfortunately progress to date has been limited and the FDA has yet to recognize any efficacious therapy for these indications. However, advances in this area including regenerative medicine coupled with novel drug delivery approaches give hope for the emergence of truly protective and restorative therapies in the near future. This article provides a brief overview of this emerging field and highlights key regulatory guidance and recommendations for preclinical assessment.
Therapeutic approaches
Historically, the primary therapy to address auditory deficits has been the use of medical devices such as hearing aids and cochlear implants. Although these devices may improve the lives of patients, they are relatively costly, sometimes inefficient in noisy environments, and do not treat the cause. More recently, there has been increased interest in pharmaceutical therapeutic targets in the auditory field, potentially expanding the market for auditory indications and allowing new entrants into this underserved clinical space. The field of audiology has long suffered from a lack of breakthroughs in disease biology and drug delivery techniques, as was the case for ophthalmology until relatively recently. Ophthalmology’s growth trajectory over the last few years has resulted in a multi-billion dollar ophthalmic drug market; the market for pharmaceutical treatments for ear disorders could easily follow the same path.
Many forms of hearing loss are mediated by the death of hair cells and the subsequent loss of synapses connecting hair cells to auditory nerve fibers in the inner ear. The biological and molecular mechanisms involved in this sensory cell death are a topic of a great deal of recent research, spurring an increase in industry investment in an effort to reach this large, almost entirely underserved market. Furthermore, molecular aspects of induced hearing pathology are recapitulated in other physiological pathways, presenting exciting opportunities for drug repurposing.
The techniques and methods used in audiology require state of the art technology and a broad and deep expertise in neurosciences and biology. Research laboratories serving this therapeutic area must be able to combine: sophisticated surgical approaches for specific otic administration, samplings of the perilymph, electrophysiology, histology, and specific expertise in analysis and interpretation.
Regulatory guidance
As with any other therapeutic treatment, entry into the clinical phase is predicated on successfully navigating regulatory requirements for safety and efficacy in appropriate preclinical models. The non-clinical studies are intended to provide proof of a positive benefit/risk ratio prior to human administration. The benefit is based on efficacy studies, called primary pharmacodynamics or specific pharmacology. For risk evaluation, the design of non-clinical safety studies must conform to the exploratory clinical trials and the collected data should anticipate potential side effects in humans. The risk is assessed in toxicology and safety pharmacology studies.
When compared with other targeted sensory indications, such as ophthalmology, the lack of regulatory specificity and historical data on assessments of auditory toxicity leave a small number of relevant sources as paramount in designing and performing a preclinical program.
The key questions for a safety package are:
- What type of studies are required?
- What are the endpoints for assessing auditory functions?
To answer the first question, the main non-clinical guideline is: The M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals – Jan 2010 – ICH. This document defines the type and duration of non-clinical safety studies and their timing to support the conduct of human clinical trials and marketing authorization for pharmaceuticals. The design of the required study needs to be customized to provide the appropriate data for evaluation by regulatory agencies.
Recommended non-clinical studies to support a single dose clinical study at sub-therapeutic doses or into the anticipated therapeutic range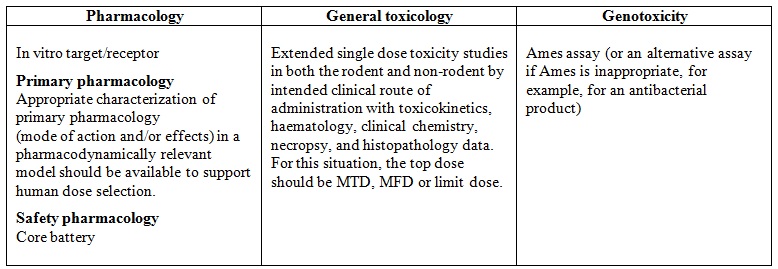
In addition, for question 2, and for the otic route specifically, we can refer to the Non-clinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route. Oct 2015 – FDA. This guideline defines the main readouts for assessing the auditory function in toxicology studies by the otic route: the auditory brainstem response (ABR) and the cytocochleogram. “For drugs intended to reach the inner or middle ear, toxicity studies should also include the evaluation of the auditory brainstem response as well as the evaluation of microscopy of relevant otic tissues including a cytocochleogram.”
The ABR measure is the most common test in non-clinical studies as well as clinical situations. The measure is objective, non-invasive and translational. Otoscopic macroscopic evaluations of the tympanic bulla (tympanic membrane and middle ear) provide additional information on outer and middle ear anatomy and pathology.
Preclinical development in the hearing field is as fascinating as it is complex. The quality and success of preclinical trials requires accurate and reproducible ABR measurements using state-of-the-art equipment in acoustics and electrophysiology, as well as high throughput and excellent technical skills. Importantly, automatic detection of ABR thresholds eliminates the variability and subjectivity associated with visual estimation. In addition, a large range of historical data provides robustness to the model, which is essential for guaranteeing the validity of analysis results.
Definition of the 2 main read-outs used for the assessment of auditory function in toxicology studies
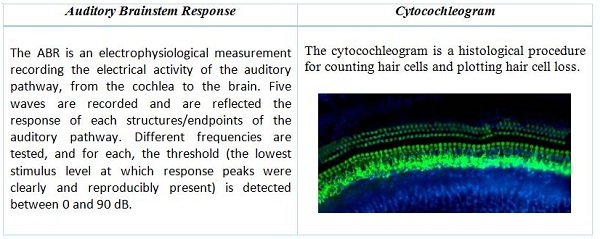
Cochlea extraction and mounting on slides requires outstanding dexterity and technical expertise. Sophisticated microscopy is necessary to deliver high-quality imaging for accurate quantification of hair cell density and distribution. State-of-the-art technology enables automatic counting, delivering significantly more accurate and consistent results in a cost-effective manner.
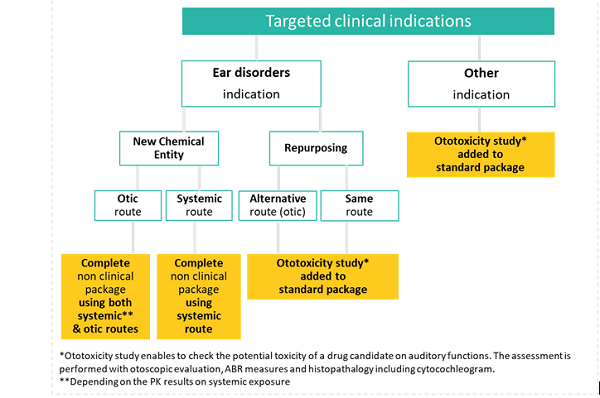 Scope of toxicology studies package according to clinical indication
Scope of toxicology studies package according to clinical indication
The middle and inner ear contain particularly delicate and difficult to access structure, and as a result, otic route delivery requires a high level of expertise and surgical skill. Moreover, due to anatomical variability, administration and surgery procedures must be adapted to each species.
The decision tree for non-clinical auditory assessments is dictated by the target clinical indication, the type of molecules (NCE versus repurposing), and the route of administration (otic versus systemic) and is summarized below (Fig. 4). At early stages, the local tolerance study can be confined to clinical signs, and macroscopic and microscopic examinations of the application site during general toxicology. Finally, systemic exposure data (ICH S3A, Ref. 7) in the species used for repeated-dose toxicity studies should be evaluated before initiating human clinical trials.
Later in the development, and specifically for the otic route, dermal irritation and delayed contact hypersensitivity must be evaluated due to the decreased thickness of the skin of the pinnae, as part of a toxicological assessment. Additionally, the ability of the test article to penetrate an intact tympanic membrane should be assessed, and the focal exposure to the middle and inner ear should be determined in models of both intact and fenestrated tympanic membranes.
Conclusion
The lack of strong historic regulatory guidelines and the technically challenging methods required to assess hearing function and appropriate anatomy render preclinical program design an especially challenging consideration when targeting auditory-related indications. Considering these critical issues early in your program development and partnering with experts in regulation and preclinical study design will maximize your chances at a successful development cycle while minimizing scientific and regulatory risk.




{kind=link}