

















Key Takeaways:
- The multi-vendor model in biopharma CMC creates communication risk, timeline gaps, and regulatory complexity
- Integrated CDMO partnerships consolidate development and manufacturing under a single organization with end-to-end knowledge of the process
- Single-partner models improve technology transfer efficiency and reduce CMC data inconsistency
Walk through the CMC timeline of a mid-stage biologic program that entered the clinic five years ago and you will likely find a familiar story. Cell line development was handled by a specialist CRO in one geography. Process development was done in-house or with a second vendor. GMP manufacturing for Phase I material went to a third organization, one that received the process documentation, reviewed it, asked clarifying questions, and then spent weeks or months getting the process to run consistently in their facility. The fill-finish work went somewhere else again.
Each of those organizations did reasonable work. Each handoff was managed professionally. And the program still lost four to six months on transfers, reconciling inconsistencies between process data generated in different environments, and answering regulatory questions that arose because the IND package told a story assembled from multiple sources rather than a single coherent development narrative.
This is the multi-vendor model. And while it remains common, it is increasingly recognized for what it actually is: a risk structure that compounds at every handoff, exactly at the moments when programs can least afford it.
How the Multi-Vendor Model Became the Default
The fragmented approach to biopharma CMC did not emerge by design. It emerged by necessity. In the early days of the biologics industry, no single organization could credibly offer cell line development, upstream process development, downstream purification optimization, analytical development, and GMP manufacturing under one roof. The specialized expertise required for each stage was genuinely rare, and companies assembled the best available partners for each function independently.
That landscape has changed significantly. Over the past fifteen years, several CDMOs have invested heavily in building genuinely integrated platforms that span the full development and manufacturing lifecycle from initial cell line establishment through commercial supply. The capability that once required four or five specialist vendors can now be accessed through a single organization with institutional knowledge of the molecule, the process, and the regulatory history.
Despite this, many biotech programs continue to operate on fragmented models partly out of habit, partly because individual vendors are selected on the basis of specific technical capabilities without sufficient consideration of the cumulative cost of integration across the full program.
The Real Cost of Handoffs
To understand why integrated CDMO partnership has become a strategic priority for well-run programs, it helps to look at what actually happens during a technology transfer between organizations.
When a biologic process is transferred from a development organization to a GMP manufacturing site, even within a contractual relationship designed to make that transfer smooth, several things reliably happen. The receiving organization conducts a technical review of the process documentation and identifies gaps or ambiguities. These require responses from the originating organization, which takes time. If the originating organization has since moved on to other programs, response quality and speed deteriorate. Equipment differences between sites require process adjustments. Those adjustments require qualification runs. Those runs produce data that may differ from the originating development data, which in turn requires scientific justification in the regulatory submission.
This process takes time in the best case. In the worst case, it produces process performance data that is inconsistent with what regulators will expect based on the development-stage data already in the file. Resolving that inconsistency can delay an IND filing by months.
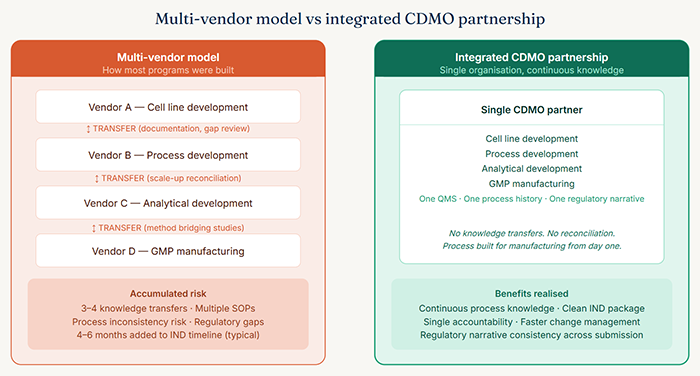
An integrated CDMO partnership eliminates or substantially reduces this specific class of risk. When the organization that developed the process is the same organization that runs the GMP manufacturing campaign, the process knowledge does not transfer between institutions it stays within the same system, held by the same people, executed in an environment that was designed with manufacturing in mind from the start.
What Regulatory Agencies Actually Want to See
FDA and EMA reviewers reading a CMC package for an IND or a BLA are evaluating one thing above all others: evidence that the applicant understands their manufacturing process well enough to control it reproducibly and to produce a drug substance of consistent quality.
A CMC package assembled from data generated across multiple organizations, on different equipment, under different SOPs, and with multiple handoffs in between, inherently carries more noise. Data sets generated under different conditions require more bridging studies, more comparative analytics, and more regulatory justification. Reviewers issue information requests not out of arbitrariness but because they genuinely cannot assess process control from a fragmented package.
By contrast, programs developed under an integrated CDMO model can present a CMC narrative that is genuinely continuous. The cell line that was selected in early development is the same one used in GMP. The process parameters established during process development were refined, not reinvented, for manufacturing scale. The analytical methods are consistent throughout because they were developed and validated within the same quality management system.
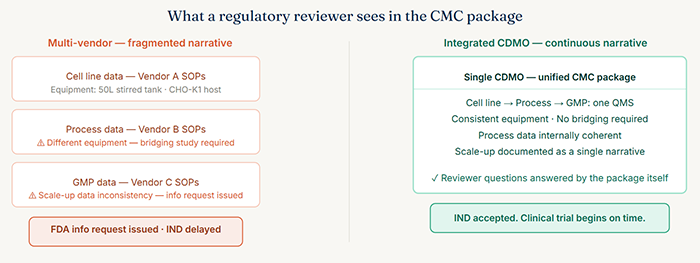
This continuity is not just cosmetically appealing to regulators. It is substantively meaningful. It demonstrates exactly the kind of process understanding that regulatory reviewers are trained to look for.
The Program Manager’s Perspective
For biopharma project managers responsible for CMC timelines, the integrated model changes the risk calculus in important practical ways.
Milestone accountability becomes cleaner. In a multi-vendor model, a delay at one vendor creates a cascade that is difficult to attribute, difficult to communicate to stakeholders, and difficult to recover from because the recovery depends on another external organization’s capacity and responsiveness. In an integrated model, the single CDMO partner is accountable for the full timeline. Delays are visible earlier. Recovery options are within the same project management structure.
Change management is faster. When a process change is required, and in early-stage development, process changes are routine, the change does not need to traverse organizational boundaries. The development team and the manufacturing team are part of the same organization, operating under the same change management system. Decisions that take weeks in a multi-vendor model can take days in an integrated one.
Resource planning is more predictable. CDMOs offering integrated development and manufacturing platforms can allocate resources across the full program timeline rather than optimizing for a specific functional scope. This reduces the capacity planning gaps that often emerge when one vendor completes their scope before the next vendor is ready to begin.
Choosing the Right Integration Model for Your Program
The shift toward integrated CDMO partnerships does not mean every program should immediately consolidate everything into a single vendor. The selection decision depends on several factors that are specific to the program, the molecule, and the stage of development.
Early-stage programs with significant scientific uncertainty, where the development path is genuinely unclear, may benefit from retaining flexibility across vendors until the program direction is established. The cost of that flexibility is real, but so is the cost of committing to a full integrated partnership before the program is mature enough to define what it needs.
Programs that have completed proof-of-concept biology and are moving toward IND filing are typically at the right stage to consolidate. By this point, the molecule is selected, the broad development strategy is defined, and the primary risk to the program is execution, timelines, regulatory submissions, GMP readiness. All of these risks are reduced by integration.
CDMOs like Lonza, Samsung Biologics, WuXi Biologics, and others have built genuinely end-to-end platforms that span from cell line development through commercial manufacturing. Each has different strengths in terms of modality coverage, platform technologies, and geographic footprint. What they share is the ability to hold process knowledge continuously across the full development lifecycle, and that continuity is increasingly what differentiates fast programs from slow ones.

The Founder’s Argument for Integration
For biotech founders, the integrated CDMO model makes a compelling argument from a different angle entirely, the investor and business development angle.
A program with a single integrated manufacturing partner presents a cleaner story. The CMC risk narrative is simpler: one organization, one process history, one accountability structure. When that partner also offers a royalty-free expression system and an established regulatory track record, the financial and regulatory risk profile of the program improves materially.
In due diligence conversations, the ability to say that development and manufacturing are being handled by a single partner, with continuous process knowledge from cell line selection through Phase I material, reduces the number of open questions an investor or potential partner needs to answer. It does not eliminate program risk. But it removes a specific category of execution risk that sophisticated investors have learned, through painful experience, to worry about.
Building a biologic drug is hard enough when everything works. The programs that reach the clinic fastest are the ones that design out the avoidable risks early, and fragmented manufacturing is one of the most avoidable risks in the modern biopharma landscape.

{kind=link}