

















At the discovery and early stages of drug development, the aim is to evaluate the pharmacology, pharmacokinetics and toxicology of a compound using a simple liquid formulation.
During this process, drugs must dissolve at a concentration high enough to allow therapeutic concentrations of the drug to reach its target. However, low aqueous solubility can prove to be a major obstacle in the development of liquid formulations. Consequently, compounds need to be formulated with excipients that can solubilise them.1/2
In selecting excipients which are able to solubilise a particular compound, the industry currently relies on a trial and error-based approach.This presents a prime opportunity for the application of a more intelligence-based methodology to formulation.
Approaches to enhance solubility
Methods to improve solubility can be categorised into a number of different techniques. These include chemical modifications (prodrug, salt formation), physical modifications (cocrystal, solid dispersions, nanocrystals), carrier systems (liposomes, emulsions, cyclodextrins) and solvent modifications (pH adjustment, co-solvent, wetting agents)3/4.The most appropriate method will depend on several factors including an API’s chemical properties, physical state of the formulation, and route of administration.In the creation of liquid formulations, solvent modifications and carrier systems are most commonly used because they affect only the solvation characteristics of a drug rather than its solid-state properties.5  Despite there being an abundance of techniques available,trial and error is the most commonly adopted approach in selecting formulation excipients to solubilize drugs.This empirical way of doing things is not only time-consuming, but also requires a large amount of materials to be used. Although successful in the long-term, for drug developers, it proves to be an extremely costly approach.A more rational approach at this stage demands a technology or methodology that can identify the best possible formulation in a more efficient way.
Despite there being an abundance of techniques available,trial and error is the most commonly adopted approach in selecting formulation excipients to solubilize drugs.This empirical way of doing things is not only time-consuming, but also requires a large amount of materials to be used. Although successful in the long-term, for drug developers, it proves to be an extremely costly approach.A more rational approach at this stage demands a technology or methodology that can identify the best possible formulation in a more efficient way.
Establishing a high throughput screen for liquid formulations
To overcome the challenges associated with traditional techniques, a new high throughput screen method has been developed to provide a more efficient way of screening an excipient’s solubilisation capacity. From the outset of the project, the aim was to establish a methodology that would use minimal amounts of API, while providing a cost effective and efficient way to achieve results. It was also vital that the platform could provide conclusive information about a compound’s stability in varying solvents and excipients.
In establishing the methodology, several experiments were performed to optimise the approach and achieve the ultimate objective of creating a robust, automatic platform. After the method had been optimised, the screening list involved a diverse range of excipients with different solubilising mechanisms, for instance6 : water-soluble organic solvents, non-ionic surfactants, water-insoluble lipids, organic liquids/semi solids, cyclodextrins and phospholipids.
Different excipients will be most applicable for different delivery systems. For example, there will be variance between those which can be classified for an oral excipient and those which are most suited to an injectable. There is also a need to understand exactly how far a concentration can go for each excipient and each drug delivery system. However, this should always be within the Generally Recognized As Safe(GRAS) list of recommended concentrations.Selecting the correct excipient, or mixture of excipients, is important becausefor example too highdose could cause pain, hemolysis and inflammation, upon injection. Therefore, insightin to drug solubility in numerous excipients could assist in selecting a formulation that minimises unwanted effects and increases patient safety.
A new methodology
The newly established high throughput screen platform is based on identifying the solubilisation capacity of each excipient for a compound to rationally select optimum excipients to make liquid formulations. It has been designed to shorten the time necessary to identify which excipients can solubilise a drug and which excipients a drug can remain stable in. Instead of testing every excipient manually, as is the case with traditional methods, this methodology enables multiple tests to be undertaken simultaneously.
The method was initially developed using six commercially available drugs with diverse chemical properties. Testing was conducted using 30 excipients dispensed in 96 well-plates via a fully automated TECAN-robotic system. The plate was shaken for at least 48 hours to achieve equilibrium.
The solubility results were compared with solubility measurements performed using a manual shake flask method where 15mgof powder and 2mL of excipient were added. The samples were again shaken for 48 hours, centrifuged, and thenanalysed by high-performance liquid chromatography (HPLC) to determine the solubility of the compounds in the excipient and detect any degradation.
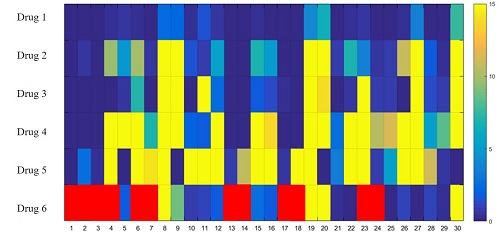
Findings
Shows the solubilisation extent for each molecule by each excipient.Though some excipients show better solubilisation capacity than others; the trend varies between the compounds. Characterising an API based on these specific molecular properties enhances the selection of the most appropriate formulation strategy, bringing better optimisation for drug delivery systems.
pH dependent solubility is a very useful approach for ionisable compounds, especially if it can be combined with another solubilising excipient. The contribution of solid-state barrier to solubilising a compound appears to be more pronounced ata cut-off level of solid state properties. This has already been determined to some extent by previous research programmes,7 and is currently being investigated using a larger set of compounds. Before this cut-off, the solubilisation of the compound was more compound specific which creates the need to also test on a larger set of excipients.
The results of the high throughput screen method have demonstrated that solubility using this technique is not statistically different (at 95% confidence interval using a t-test) to that achieved when using a manual approach. The method can provide formulation scientists with detailed information on the solubilisation capacity of different excipients for different compounds, while also offering insight into the stability of the compound in an excipient. Additionally, it can provide greater understanding of the solubility and stability of a drug in water, and acidic and basic buffers.
The high throughput screen method overcomes many of the challenges associated with manual approaches by being more cost-effective and economical in the use of materials. The 3-5-day turnaround time for results (per set of compounds) is also significantly faster than traditional approaches.
Final thought
Screening processes for potential drug candidates require liquid formulation that is suitable for injection. However, the solubility of compounds is regarded as one of the most difficult obstacles in successful formulation development. Establishing a new high throughput screen platform has not only created new potential for reducing costs, but has opened-up new possibilities in improving timelines and the probability of successful formulation. By simplifying formulation design, a faster and more effective roadmap for the development of potential new drugs can be achieved.
References
1.Li, P., & Zhao, L. (2007). Developing early formulations: practice and perspective. International Journal of Pharmaceutics, 341(1), 1-19.
2.Dahan, A., Beig, A., Lindley, D., & Miller, J. M. (2016). The solubility–permeability interplay and oral drug formulation design: Two heads are better than one. Advanced drug delivery reviews, 101, 99-107.
3.Alhalaweh, A., Bergström, C. A., & Taylor, L. S. (2016). Compromised in vitro dissolution and membrane transport of multidrug amorphous formulations. Journal of Controlled Release, 229, 172-182.
4.Miyako, Y., Khalef, N., Matsuzaki, K., & Pinal, R. (2010). Solubility enhancement of hydrophobic compounds by cosolvents: role of solute hydrophobicity on the solubilization effect. International journal of pharmaceutics, 393(1), 48-54.
5.Alhalaweh, A., Roy, L., Rodríguez-Hornedo, N., & Velaga, S. P. (2012). pH-dependent solubility of indomethacin–saccharin and carbamazepine–saccharin cocrystals in aqueous media. Molecular pharmaceutics, 9(9), 2605-2612
6.Strickley, R. G. (2004). Solubilizing excipients in oral and injectable formulations. Pharmaceutical research, 21(2), 201-230
7.Persson, L. C., Porter, C. J., Charman, W. N., & Bergström, C. A. (2013). Computational prediction of drug solubility in lipid based formulation excipients. Pharmaceutical research, 30(12), 3225-3237.




{kind=link}