



















Anticipate New Compliance Standards Now or Risk Falling Behind
Overview
- While showing significant promise, regenerative medicine materials are inherently fragile and more at risk than other products during the supply chain process. Cross-contamination, orientation and handling problems, and equipment failure all have potential to damage irreplaceable materials.
- To avoid this, traceability, along with efficient data management, is imperative and can be addressed using Chain of Compliance™ processes. This efficiently traces the equipment and processes used in managing the environmental control of the product.
- While few industry-wide standards currently exist, the industry can expect new regulations in nine areas, which are outlined in this white paper.
- Ensuring compliance, however, can be a lengthy endeavor. This white paper outlines seven steps that companies should take now to ensure future compliance.
- Companies who fail to anticipate compliance standards now will fall behind.
Significant Changes in Regenerative
The advent of regenerative medicine is among the most significant events in modern medicine. The first large-scale launches of these therapies took place in 2017,1 and a large range of cell therapies, therapeutic tissue engineering products, human cell and tissue products, and therapies or products that combine some of these techniques are currently under development around the world.
As of Q3 2018, there were more than 1,000 clinical trials underway utilizing multiple variants of regenerative medicine technology. More than $20 billion has been invested in regenerative medicine in the last three and a half years alone, with a strong focus on oncology.2 In addition, significant mergers and acquisitions have taken place in this market.
In 2017, Gilead acquired Kite Pharma for $11.9 billion and Johnson & Johnson entered the CAR-T field with a $350 million Legend Biotech deal. In 2018, Novartis acquired AveXis for $8.7 billion3 and Celgene Corporation acquired Juno Therapeutics for $9 billion. Lastly, Bristol-Myers Squibb announced in January 2019 its intent to acquire Celgene in a cash and stock transaction with an equity value of approximately $74 billion.
Regenerative medicine Phase III clinical trials are up 37% year over year.4
The biopharmaceutical market is starting to react to the regenerative medicine opportunities, in many cases playing catch-up due to the speed in which these therapies are moving through the clinical pipeline. This speed is in part due to the passing of the 21st Century Cures Act, which led the FDA to create the Regenerative Medicine Advanced Therapy or “RMAT” designation
The act, intended to speed approval of important new treatments by leveraging novel clinical trial designs and real-world evidence (RWE), is having the desired effect.5 To further help the development of new gene therapies, in the spring of 2018, the FDA released disease-specific guidance documents for these therapies.6 The Massachusetts Institute of Technology predicts 15 to 30 gene therapy products will be approved by the FDA by the end of 2024,7 derived from a pipeline of more than 1,000 as of Q3, 2018. Furthermore, clinical progression for personalized therapies for diseases from hemophilia to cancer is now happening in as few as four years.
As described in Section 3033 of the 21st Century Cures Act, a drug is eligible for regenerative medicine advanced therapy (RMAT) designation if:
- The drug is regenerative medical therapy as regulated under Section 361 of the Public Health Service Act and 21 CFR Part 1271, Code of Federal Regulations
- The drug is intended to treat, modify, reverse, or cure a serious or life threatening disease or condition;and
- Preliminary clinical evidence indicates that the drug has the potential to address umet medical needs for such or condition.
The opportunity is large. But so are the challenges.
Because of the speed at which these therapies are developing, companies are struggling with a wide range of challenges, from integrating new manufacturing techniques, to answering bioethics questions, to ensuring product safety and efficacy, and more. Executives seeking a proven playbook for how to manage all of these issues will not find one: While industry wide standards are urgently needed, few exist.
Organizations like the Alliance for Regenerative Medicine (ARM) and the Foundation for the Accreditation for Cellular Therapy (FACT) are stepping up to address this challenge. They have instituted review bodies to analyze all aspects of the collection, manufacture, transportation, and administration of regenerative medicines.
A substantial effort is also underway at the Standards Coordinating Body (SCB), which also was established by industry and academic institutions to provide regulatory guidance for standardization of cell and gene therapy process workflow. The ISO/AWI 21973 working group, established by the SCB, includes 28 individuals representing two government institutions, three membership organizations, and no less than 19 industry organizations. The SCB has drafted documentary standards, one for cell therapy suppliers and one for transportation and logistics companies to “define minimum requirements for regenerative medicine information technology infrastructure and transportation, shipment tracking and monitoring protocols, chain-of custody requirements, and centralized logistics management systems.”8 Given the critical need to ensure quality across the entire chain, the SCB is wisely coordinating the combination of these two standards into a single standard.
The unique sensitivity of regenerative medicine products creates an entirely new set of supply chain imperatives.
Regenerative medicine products are significantly more fragile and costly than tablets or pills, and are in most cases, irreplaceable. This means that emerging product safety and efficacy standards must consider the entire supply chain, including temperature-controlled distribution. The miracles performed in labs and manufacturing mean little if the products are unusable by the time they arrive. What’s more, a casual visual inspection is of no use. If regenerative medicine products are damaged in shipment, it’s not obvious; they don’t change in smell or color. The only way to be certain if the therapy remains viable is through a constant flow of data.
 “We’re at a key point when it comes to cell and gene therapy. These therapies have the potential to address hundreds, if not thousands, of different rare and common diseases. For a long time, they were largely theoretical constructs. Now they’re a therapeutic reality. And it’s my expectation that they will soon become the mainstay of how we treat a wide range of illness.”
“We’re at a key point when it comes to cell and gene therapy. These therapies have the potential to address hundreds, if not thousands, of different rare and common diseases. For a long time, they were largely theoretical constructs. Now they’re a therapeutic reality. And it’s my expectation that they will soon become the mainstay of how we treat a wide range of illness.”
Scott Gottlieb, M.D., Former Commissioner of the U.S. Food and Drug Administration”
Precision medicine requires end-to-end precision traceability: everything from chain of custody, to chain of condition, to chain of identity, and Chain of Compliance™.
There are many risks that a product may encounter while in transit. For example, consider these three:
- Cross-contamination due to poor cleaning procedures of transit equipment
- Orientation and handling issues due to a lack of courier training
- Equipment failure events often stemming from inadequate qualification and maintenance processes Companies need to effectively address risk during drug product distribution of regenerative therapies and must consider employing standards similar to those currently being utilized during manufacturing of these therapies under cGMP conditions.
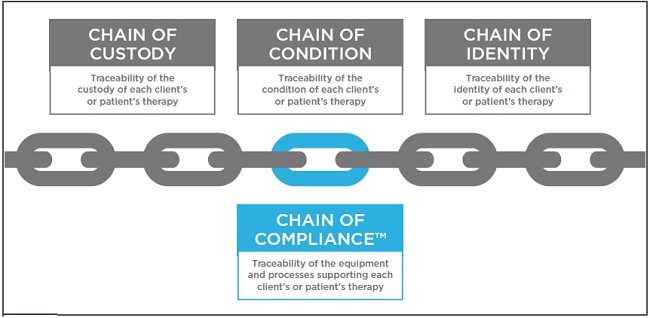
Traceability should be part of supply chain management.
The CMC’s (Chemistry, Manufacturing, and Controls) guidance in the FDA’s section of the Code of Federal Regulations (CFR) that dictate pharmaceutical manufacturing standards describe a comprehensive quality systems (QS) model, highlighting the model’s consistency with regulatory requirements for manufacturing human and veterinary drugs, including biological drug products. The CFR requires that records be made for each step in the manufacturing and distribution of products. They should be made so that any lot may be traced by an inspector at any successive step in the manufacturing or distribution process.
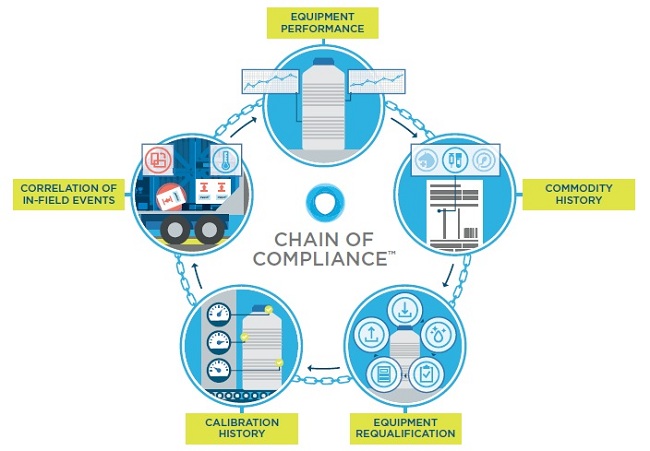
 For this reason, traceability must be a part of any long-term regenerative medicine supply chain management solution. Traceability can be addressed using Chain of Compliance™ processes, which establish full traceability of the equipment and processes used in managing the environmental control of the commodity. This includes equipment performance, requalification history, commodity history, courier handling and performance history, calibration history, and correlation competencies that can link in-field events to equipment performance.
For this reason, traceability must be a part of any long-term regenerative medicine supply chain management solution. Traceability can be addressed using Chain of Compliance™ processes, which establish full traceability of the equipment and processes used in managing the environmental control of the commodity. This includes equipment performance, requalification history, commodity history, courier handling and performance history, calibration history, and correlation competencies that can link in-field events to equipment performance.
As the number of shipments rises, the problems caused by ad hoc efforts related to shipper traceability will multiply quickly. To maintain compliance, five critical aspects must be considered:
- Equipment Performance History: Nitrogen evaporation rate (NER), vacuum integrity, and hold time data, etc.
- Commodity History: Identifying/tracking human-use-only shippers; complete records of what commodity/product has been in the unit.
- Equipment Requalification History: Comprehensive records of the deviations the package has experienced, maintenance history, damage, and contamination.
- Calibration History Data for Shipper and/or Accessories and Data Loggers: Data to ensure shippers, data loggers, and other accessories have continually operated within specified parameters.
- Correlation of In-Field Events to Shipper Performance: The ability to improve performance over time relies on knowing what happened in-field, and what the impacts are.
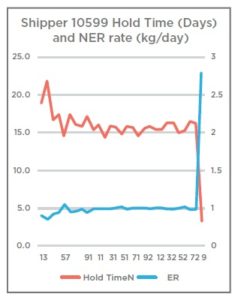
Case Study 1: Rapid unknown shipper loss of hold time due to NER failure.
Shipper 10599 had been used on a total of 29 shipments with a nitrogen evaporation rate (NER) between 0.42 kg/day and .66 kg/day, supporting a hold time of between 14.4 and 22.1 days. On return of use 29 — a trip between Amsterdam and Barcelona, Spain — the shipper underwent requalification testing and presented an NER of 2.76. This reduced the hold time of the shipper to 3.5 days despite no obvious external damage to the shipper. This shipper was removed from service immediately and scrapped.
Mitigate risk with efficient data management.
Robust data management is key to mitigating risk and better understanding variability. But perhaps most importantly, robust near-real-time data can alert you to problems before they become costly failures. In the field of personalized medicine in particular, logistics can become very complex while at the same time may require strict timing requirements and zero tolerance for temperature excursion.
While knowing the temperature of your package is essential, having comprehensive condition monitoring about your package’s pressure, orientation, humidity, shock events, and even light can alert you to potential security breaches, compromised packaging, or premature opening. This data provides the ability to assess risk and intervene when unexpected problems arise in transit, giving you the power to save time and money, while protecting what may be invaluable or irreplaceable materials, perhaps even a life-saving therapy.
A robust informatics platform to scientifically monitor and evaluate these individual impacts is imperative. Without constant monitoring, there will always be uncertainty of the efficacy or activity of the therapy upon delivery.
Where is the market headed? Nine major areas likely to be regulated.
While no one, including the FDA, knows yet precisely what the new regulatory requirements will be, today’s regulations, standards, and guidance documents provide a useful guide to the areas where logistics executives will want to focus. A robust informatics platform to scientifically monitor and evaluate these individual impacts is imperative. Without constant monitoring, there will always be uncertainty of the efficacy or activity of the therapy upon delivery.
- Quality Systems Architecture: ISO 9001:2015, EMA <2013/C 343/10>, ICH Q10, and WHO Annex 5 all point to the need for a comprehensive, quality systems approach to the distribution of regenerative therapies. While these standards were established for the development and manufacture of pharmaceutical drug substance and drug product, it makes sense that the same basic standards will apply to supply chain systems, equipment, and logistics management. Q10 embraces a risk-based approach to quality, which marries well with Quality by Design (QbD) elements related to risk management via equipment performance as well as in-field logistics.
- Quality Systems Design: ICH Q8-10 are complementary to one another and provide a foundation for the design of a quality system based on a QbD and Process Analytical Technology (PAT) methodology.10 This approach, which helps address multidimensional external impacts on product integrity, is well suited for managing risk related to complex logistics and in-field risk management.
- QbD, Risk Management: Automating risk management through QbD using real-time informatics is important when delivering commercial regenerative medicine product. Informatics to capture, interpret, and predict, as well as to provide automated alerts and pre-defined escalation protocols based on the nature of the data captured for hundreds, if not thousands, of concurrent shipments will be critical in the effective launch of these therapies. QbD processes provide the foundation enabling such an approach to be effective.11
- Document Control: ISO9001:2015, EMA <2013/C 343/10>, 21 CFR Part 11, ICH Q10, and WHO Annex 5 provide an effective framework for the establishment of:
- A records management system
- The controls associated with data collection and management
- Smart, predictable business practices
- The criteria under which electronic records can be considered accurate, authentic, and non-alterable.
5. Electronic Equipment and Software Calibration/Validation: In addition to effective records management, ICH Q7 also dictates the calibration (5.3) and validation (5.4) of the equipment and software utilized in the collection of temperature, location, and equipment traceability, key to Chain of Compliance™ and QbD processes.12.
- Equipment Validation and Traceability: Regenerative medicine requires end-to-end traceability:
everything from chain of custody, to chain of condition, to chain of identity to Chain of Compliance™, which, as mentioned above, is the traceability of the equipment, processes, and logistics handling used in managing the environmental control of the therapy while it is in transit.13 21 CFR 210, 21 CFR 211, EMA <2013/C 343/10>, and ICH Q7 provide the core components that need to be considered in the establishment of a Chain of Compliance™ competency in support of regenerative medicine distribution. Specifically, CFR 211.67 equipment regarding equipment cleaning and maintenance should be considered.
- Facilities and Personnel: Relevant Good Manufacturing Practice (GMP) standards in support of clinical and commercial product distribution related to personnel and facility management, equipment cleaning, storage and maintenance, as well as supplier and materials management, are covered under 21 CFR 210, 21 CFR 211, and EMA <2013/C 343/10>. Key anticipated emerging requirements related to equipment maintenance and traceability in support of Chain of Compliance™ standards are covered specifically in 21 CFR 210 and 211.
- Distribution and Logistics: USP <1079> is a key minimum consideration in the distribution and storage of drug product supported additionally via EMA <2013/C 343/10>, WHO Annex 5, as well as ISTA 3A,7D, and 7E. These regulations support the basic temperature and logistics management requirements including, but not limited to, equipment performance, temperature management, and lane qualification and validation. These should be augmented with a QbD methodology to manage in-field risk due to third party handling and — at a minimum — need to include active temperature, equipment, and location monitoring.
- Contamination and Packaging: Another newly emerging consideration related to cross contamination specific to cell and gene therapies is supported by EMA <2013/C 343/10>, 21 CFR 211.67, and 21 CFR 1271.190. Consensus is emerging that all equipment utilized in the distribution of blood and cell materials and final product must be transported in equipment verified as having been only ever used to transport human clinical and commercial product. This is important in order to minimize contamination risk due to non-human commodities.

There are seven steps to take now to ensure future compliance.
- Be proactive.
Given the rapid pace of change, it’s not surprising that some companies are not entirely prepared to respond. However, the longer your company waits to make progress, the more intense the stress on your organization will become and the further you will fall behind competitors. Position your company now to take maximum advantage of the growth ahead.
- Start educating your colleagues immediately.
Educate yourself and your colleagues about the lengthy process your company faces if the new requirements force you to change vendors. Even if you move aggressively, changing vendors typically takes between six and 12 months. Among other things, your company will need to:
- Re-qualify the vendor
- Re-run all shipping validations with the different format
- Conduct a series of shipping studies with saline over the course of several months
- Research, evaluate, and meet one or more vendors to review their validation documentation and complete a form alaudit of their systems and processes. While the FDA will grant companies a brief grace period for compliance, the time that is lost will be lost forever. Companies that decide to “wait and see” will find themselves significantly far behind competitors that took a proactive approach.
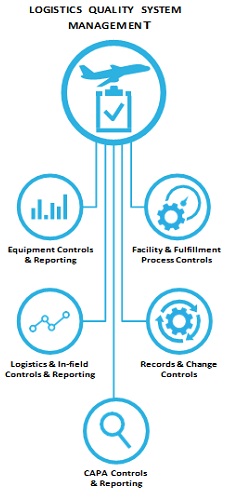
- Begin taking a six-systems quality approach to regenerative medicine distribution.
The six-systems quality approach used by many companies in pharmaceutical manufacturing can and should be adapted to regenerative medicine distribution.
Logistics Quality System Management is at the core, with five subsystems that must work together to consistently achieve compliance:
- Facility and fulfillment process controls
- Records and change controls
- CAPA controls and reporting
- Logistics and in-field controls and reporting
- Equipment controls and reporting
The performance of each these subsystems and their ability to work together effectively are heavily reliant on timely and precise data. While your quality systems may be sufficient for the more forgiving task of transporting tablets, you will almost certainly need to make some modifications within all five of these subsystems to meet the demands of the regenerative medicine market.
As one example, transporting regenerative medicines requires informatics that can properly correlate third-party effects including shock, temperature excursion, dewar orientation, and more. Trusting third-party, bolt-on informatics systems with non-indexed data streams to packaging and logistics can be a costly mistake — and no corrective action is possible once a cell-based therapy has been destroyed in transit.
- Benchmark existing vendors against potential new regulatory requirements.
How well or poorly would your existing vendors perform if new requirements were instituted tomorrow? Are they currently performing at a level well above existing regulations? Have your vendors been proactive, and are they prepared for changing regulations?
Where are the gaps in their approach, and what are their plans to address those? Remember that brand new capabilities, even from a vendor that has been reliable in the past, take time to be tested in the real world.
If your vendors aren’t ready, you won’t be either.
- Review your internal supply chain to determine if you have traceability within your logistics and distribution.
Determine if your existing supply chain has compliance traceability established. If not:
- What gaps exist?
- How critical are those gaps?
- Where do critical and non-critical gaps exist, and what other parts of the chain could they potentially impact?
- Critically evaluate existing shipping validation to determine if it sufficiently addresses the risk profile for the existing commodity.
Optimal qualification of shippers is executed by using a two-pronged approach. First, prior to any new product
launch, shippers are tested according to various standards developed to ensure the shipper meets thermal performance standards, shipping and handling standards, and infectious substance regulations. The following testing standards are used to perform this qualification:
Infectious substance shipping
- Regulation / guidelines:
– IATA Packaging Instruction 650 (Class 6 – Toxic and Infectious Substances UN 3373
Holding time performance
Regulation/guidelines:
– ISTA 7E-2010 (Heat) and ISTA 3A-2008 (Thermal and Performance Validation)
Shipping and handling performance
- ISTA 3A Transit Tested Certification
The second part of the qualification process is performed after product launch. Each shipper is requalified after every shipment to verify it will be capable of meeting shipment requirements for the next order. This requalification consists of thermal performance testing, cleaning using a validated cleaning process, and holding time analysis.
Ongoing shipper requalification
- Nitrogen evaporation rate test
– Determines the thermal efficiency of the shipper
LN2 capacity test
– Determines the amount of LN2 that can be absorbed in the shipper
Dynamic holding time analysis
– Uses the prior test result to determine the approximate dynamic holding time to be used for the next shipment.
- Ensure your current supply chain processes provide sufficient visibility related to new compliance requirements. On July 11, 2018, the FDA issued six new draft guidance documents on the development of cell and gene therapy products. Three of these provide new and comprehensive updates to existing guidelines that address manufacturing issues related to cell and gene therapy. These guidance documents are likely only the starting point of the emergence of regulatory requirements that will be required in support of these therapies long-term.14
Conclusion
The future of regenerative medicine is bright. However, the accelerated pace of change means the time to begin significantly improving your company’s logistics and distribution risk management capabilities is now. Cryoport can help make the changes necessary to ensure your company achieves compliance standards in the regenerative medicine market. Intelligent, effective use of QbD and Chain of Compliance™ will enable proactive quality management processes and minimize the risks of falling behind. Compliance, not complacency, is the right strategy.
Reference
1.Novartis. Novartis receives first ever FDA approval for a CAR-T therapy. https://www.novartis.com/news/media-releases/ novartis-receives-first-everfda- approval-car-t-cell-therapy-kymriahtm-ctl019. Accessed March 11, 2019. And, Gilead. Kite’s Yescarta Becomes First Car T Therapy Approved by the FDA for the Treatment of Adult Patients With Relapsed or Refractory Large B-Cell Lymphoma After Two or More Lines of Systemic Therapy. https://www.gilead.com/newsand- press/press-room/ press-releases/2017/10/kites-yescarta-axicabtagene-ciloleucel-becomes-first-car-t-therapy-approved-by-the-fda-for-the- treatment-of-adultpatients- with-relapsed-or-refrac. Accessed March 11, 2019.
2- Outcome Capital. Cell-Based Therapies: A Maturing Market Poised for Growth. https://www.outcomecapital.com/wp-content/uploads/2018/10/Regenerative_Med_Final.pdf. Accessed March 11, 2019.
3- Chilberg M. Genomic Medicine: Catch The Gene Therapy Wave. Seeking Alpha. https://seekingalpha.com/article/4173868-genomic-medicine-catchgene-therapy-wave. Accessed March 11, 2019.
4- Alliance for Regenerative Medicine. State of the Industry. Mid-Year Update. https://alliancerm.org/wp-content/ uploads/2018/05/ARM_Janet_MidYearSOTI_FINAL.pdf. Accessed March 11, 2019.
5- Barlas S. The 21st Century Cures Act: FDA Implementation One Year Later. P&T. 2018 Mar; 43(3): 149-151, 179. https:// www.ncbi.nlm.nih.gov/pmc/articles/PMC5821241/. Accessed March 11, 2019.
6- Alliance for Regenerative Medicine. RMAT Designation: Impact on Regenerative Medicine Sector & How it Compares With Other Accelerated Approval Programs. https://alliancerm.org/wp-content/uploads/2018/06/ARM-RMAT-Webinar-Slides-PDF.pdf. Accessed March 11, 2019.
7- MIT NEWDIGS. Research Brief. 2018 Oct; 2018F210v027. https://newdigs.mit.edu/sites/default/files/FoCUS%20 Research%20Brief%202018F210v027. pdf. Accessed March 11, 2019.
8- Standards Coordinating Box. Transportation Requirements for Cell Therapy Products. https://www.standardscoordinatingbody.org/project-transportationrequirements-of-cell-therapy-products. Accessed March 11, 2019.
9- U.S. Food and Drug Administration. Remarks by Commissioner Gottlieb to the Alliance for Regenerative Medicine’s Annual
Board Meeting. https://www.fda.gov/NewsEvents/Speeches/ucm608445.htm. Accessed March 11, 2019.
10- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.ICH Harmonised Tripartite Guideline: Pharmaceutical Development Q8(R2). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline. pdf. Accessed March 11, 2019.
11- Sawicki M. The Arrival of Quality by Design in Logistics. World Pharma Today. http://www.worldpharmatoday.com/Articles/ the-arrival-of-quality-by-designin-logistics. Accessed March 11, 2019.
12- U.S. Food and Drug Administration. Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients,Guidance for Industry. https://www.fda.gov/downloads/Drugs/…/Guidances/ucm073497.pdf. Accessed March 11, 2019.
13- Sawicki M. The Chain of Compliance. Contract Pharma. https://www.contractpharma.com/contents/view_ features/2018-06-12/the-chain-of-compliance/. Accessed March 11, 2019.
14- U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D., on agency’s efforts to advance development of gene therapies. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm613026.htm. Accessed March 11, 2019.



{kind=link}