


















As of January 31, 2022, the EU has implemented the Regulation 2014/536 on clinical trials for Medicinal Products (MPs) intended for human use. This regulation provides a centralized approach for electronic submission and evaluation of clinical trials. Alongside this, the field of Medical Devices (MDs) has also experienced a significant technological advancement through EU Regulation 2017/745. Within this field, the market for Substance Based Medical Devices (SBMDs) has been expanding, accounting for 10% of all self-medication sales across European countries in recent years.
The EU-CTD is a directive that outlines a series of objectives to be applied in individual European countries, and subsequently incorporated into national legislation across Europe. In contrast, the EU-CTR is a legally binding act that takes immediate effect across all European countries. Previously, trials that took place in multiple member states were required to submit separate applications to each state. However, the creation of a unified environment now allows for a single application to be evaluated by all participating member states.
What is the Clinical Trial Information System
The EU-CTR operates through the use of a newly developed portal database called the Clinical Trial Information System (CTIS). This online system acts as a centralized, paper-free platform that allows for the submission, review, regulation, monitoring, and reporting of all trial-related information between relevant member states. The CTIS will be managed by the European Medicines Agency (EMA), working in conjunction with the Member States and the European Commission.
The CTIS comprises three distinct user-based portals: the sponsor workspace, authority workspace, and a public website. Users with a valid EMA account can easily gain access to the CTIS workspaces.
The Vital Prerequisites for Accessing the CTIS portal
- Existing EMA account holders request access to the CTIS portal
- Register organization/trail sites in OMS (Organization Management Service)
- Ensure the medicinal products are registered in xEVMPD.
Key Benefits of Utilizing the CTIS portal
There are multiple benefits in utlising the CTIS portal
- Increased Trial Transparency: The trial process becomes more transparent to the public throughout its entire duration.
- Data Disclosure: Depending on the confidentiality agreement, sponsors may choose to disclose data to the public.
- Risk Based Trials: The EU-CTR, also known as the low intervention study, enables sponsors to conduct risk-based trials. While the applications do not expire under the EU-CTD, they will expire under the EU-CTR if no patients are enrolled within two years.
- Event Reporting: The EU-CTR requires that serious breaches be reported within 7 days and that the Trial Master File (TMF) be archived for 25 years.
Getting Authorization Right
The Regulation describes a precise procedure for the submission, assessment, and evaluation of requests for authorization of clinical trials. The application of the dossier involves two parts.
- Part 1 pertains to the general scientific or technical data that would be standardized for all member states.
- Part 2 contains the national, ethical, and local information concerned for all member states. This is assessed for its compliance with Directive 95/46
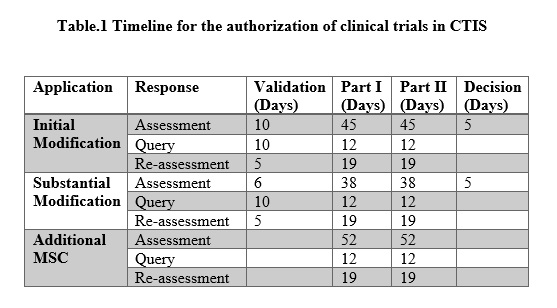
The clinical trial can be made effective within 60-91 days from the commencement of the application in the CTIS domain. The timeline is provided in table.1.
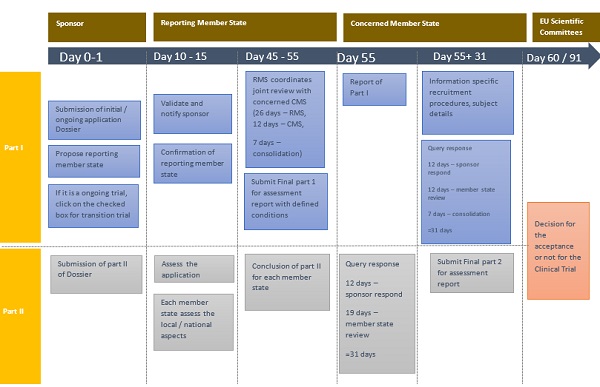
Fig.2 Authorization procedure for Initial / Transitional clinical trials via CTIS
Effective Transition to the CTIS Portal
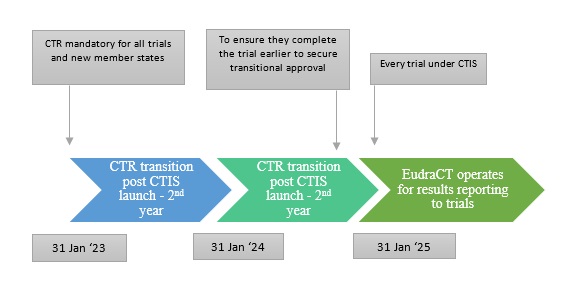
The implementation of the CTIS regulation will improve the transparency and reliability of clinical trials across Europe by ensuring that every application is aligned with both the National Competent Authorities (NCA) and Ethics Committee (EC). The focus of the CTIS transition is to ensure compliance with the transitional trials, meaning that all CT Directive trials must be registered as transitional trials in the CTIS before the January 31st, 2025 deadline.
Building a Robust Transition with a Strategic Partner
Sponsors have the opportunity to optimize their clinical trial applications and evaluation procedures throughout the European Union, while also enhancing safety standards and expediting safety reporting. To achieve this, sponsors can develop a transition strategy that addresses their specific needs and ensures a seamless integration of their application into the CTIS system.
. Fig.1 Creation of a trial in the CTIS domain
The initial trial application versus transitional trial application
The initial and the transitional trial have the same procedure for authorization. However, the transitional trial procedure differs in two aspects as follows;
- For the transitional trial, select the check box while creating a trial into the CTIS domain (Fig.1)
- The EudraCT number reference will be different for every clinical trial application
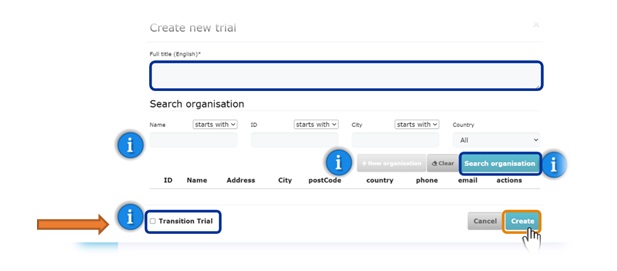
Fig.3 Transitioning period of CTIS
There have been various organizational and resource-related issues with clinical trials in the past. However, the introduction of the single portal CTIS domain has overcome these drawbacks and improved the regulation of clinical trials. The submission of a dossier is a crucial step in starting a clinical trial application (CTA), which involves significant work to comply with EU regulations. Working with expert professionals is key to ensuring a successful end-to-end clinical trial transition in the EU.
Clinical trials play a central role in the pharmaceutical research and development process, providing important information for decision-making on medicines pricing, market approval, reimbursement, and clinical practice. Therefore, ensuring clinical trial transparency is critical for various stakeholders, including policymakers, public health organizations, researchers, healthcare professionals, and patients. In the EU, clinical trial application involves various strategies and management approaches to comply with regulations. The CTIS system will harmonize all trials under one domain and streamline the regulation process.
References:
- Clinical Trials Information System | European Medicines Agency (europa.eu)
- https://www.europeanpharmaceuticalreview.com/article/168043/understanding-the-new-eu-clinical-trial-regulation-seven-things-sponsors-should-know/
- https://www.raps.org/regulatory-focus/news-articles/2017/4/impact-of-the-revised-eu-clinical-trial-regulation
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02014R0536-20221205
- https://spor.ema.europa.eu/sporwi/
- https://www.ema.europa.eu/en/human-regulatory/research-development/data-medicines-iso-idmp-standards/spor-master-data/organisation-management-service-oms
- https://www2.deloitte.com/content/dam/Deloitte/nl/Documents/risk/deloitte-nl-risk-ctr-report.pdf
- https://www.ema.europa.eu/en/documents/other/member-states-guide-transition-trials-eudract-ctis-ctis-training-programme-module-23_en.pdf
Authors
 Bhuvaneswari Sivaraman
Bhuvaneswari Sivaraman
Regulatory SME, Regulatory Operations
Navitas Life Sciences
Having attained a Master’s degree in Pharmacy, Bhuvaneswari possesses proficiency in a variety of tools, including QSAR, DOCKING Software, Visualizing Tool, Sketching, and MS Office. Additionally, she has contributed to the field of drug development research by publishing approximately six articles in various scientific journals.

Velkumar Kuzhandhaipandian
Senior Manager, Regulatory Operations
Navitas Life Sciences
With a wealth of professional experience, Velkumar specializes in regulatory affairs and excels at compiling formulation dossiers and API DMFs in accordance with the requirements of health authorities. Additionally, he is skilled at reviewing CMC documentation and ensuring regulatory compliance in adherence to the regulations of the pharmaceutical industry.
 Ragini Tushar Bhandari
Ragini Tushar Bhandari
Senior Manager, Regulatory Operations
Navitas Life Sciences
Possessing a bachelor’s degree in pharmacy, Ragini holds extensive experience in Drug Regulatory Affairs. She has been involved in numerous regulatory projects, such as compliance, Remediation, CMC authoring for Variations, renewals, and post-approval submissions.


{kind=link}