


















How an emerging mass spectrometry technique – TIMS – is revolutionising shotgun proteomics.
High-performance liquid chromatography with time-of-flight mass spectrometry (HPLC–TOFMS) has become a powerful technology for identifying and quantifying proteins. Having said that, it’s still very challenging to achieve coverage of complete proteomes due to of the limited speed, sensitivity, and resolution of current mass spectrometers.But a relatively new technique—trapped ion mobility spectrometry (TIMS)—is promising to change things, by providing another way to separate ions from each other. This article takes a look at TIMS, and what it means for studies into post-translational modifications (PTMs) using shotgun proteomics.
Separating ions by their shape
TIMS is a gas-phase mass spectrometry (MS) technique that enables ions to be separated according to their collisional cross-section (CCS)—a measure of how likely they are to be deflected by a collision with other gas molecules as they drift through an ion tube. So compact ions (with a small CCS) move faster than large ions (with a large CCS).
What’s the benefit of this? Put simply, it provides another way of separating ions—adding an additional‘dimension’ to the analysis. So, as well as separating proteins or peptides by their retention time in the HPLC system, and separating ions by their mass-to-charge ratio in the TOF MS instrument, we can now separate ions by their shape.
To understand what this means for shotgun proteomics, let’s first look at how TIMS works.
How TIMS works
The TIMS system uses a dual-tunnel setup: the first tunnel accumulates ions, ensuring that sensitivity is maximized. The ions then pass into a second tunnel, where the ions are separated and concentrated according to their mobility and from where they are sequentially released depending on their CCS, and their mass or fragmentation spectra measured. Importantly, both steps can run simultaneously,ensuring that no ions are lost. This parallel accumulation serial fragmentation approach, known as PASEF, makes it possible to achieve a duty cycle of nearly 100%, resulting in an ion loss of virtually zero (Figure 1).
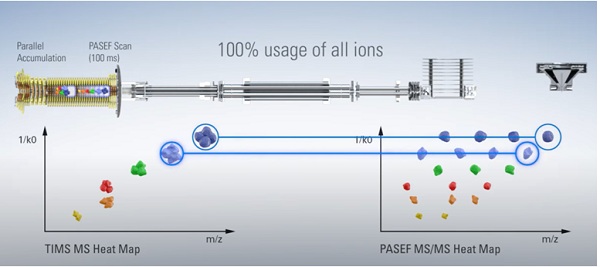
Figure 1: Generation of TOF MS heat maps using (left) parallelaccumulation TIMS technology (PASEF), and (right) the added sequencing capability possible by synchronizing the quadrupole mass window with the TIMS elution times.
After the TIMS step, ions pass into a segmented quadrupole mass filter, which lies at the heart of another advantage of TIMS, namely the ability to improve sequencing speed. This is achieved by synchronizing the quadrupole mass window with the elution times of the same ions from the TIMS analyzer. The result is that sequencing speeds of over 100 Hz can be attained without losing sensitivity or resolution.
But what does the TIMS capability mean in practice? Below are two examples from our recent work at PTM Bio.
Example: Ubiquitination
Ubiquitination is a PTM that is essential for regulating key biological processes, including protein degradation, immune response, signal transduction, and DNA repair. Unfortunately, characterizing global ubiquitination using MS is particularly challenging because of the sub-stoichiometric abundance of this PTM, and because of the co-elution of ubiquitinated peptides only differing in the position of the modified site.
To examine how TIMS could help with this challenge, we performed an in-depth characterization of ubiquitinated peptides from human cells.[1] The peptides were enriched from HeLa cell lysates with a high-affinity enrichment kit, and analyzed by LC–MS/MS with TIMS.
The results were very encouraging: more than 17,000 ubiquitination sites were identified from four HeLa cell samples with only 50-min gradients, corresponding to 5093 ubiquitinated proteins. The additional dimension of separation provided by TIMS enabled us to identify and quantify co-eluting isobaric peptides with different modification sites (Figure 2). Importantly, the high sensitivity of the TIMS method helped us identify site-specific PTM isomers with high reproducibility.
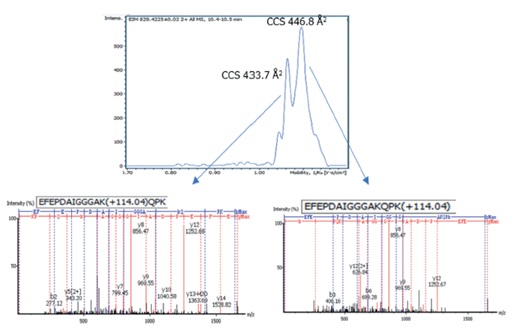
Figure 2: Separation of two co-eluting isobaric ubiquitinated peptides with different modification sites by ion mobility. Top: The extracted ion mobilogram for m/z 829.4115 at 10.45 min. The isomers can be resolved by their collisional cross-section (CCS). Bottom: Non-chimeric MS/MS spectra of the two site-specific ubiquitinated peptides.
Example: Acetylation
Acetylation is another well-known PTM that plays an essential role in cellular processes, including transcriptional regulation, cell cycles, and apoptosis. However, the low abundance and high dynamic range of acetylation make thorough MS-based profiling particularly difficult. We were therefore interested to see whether TIMS technology could help.
In our study, acetylated peptides were enriched from mouse liver and rice leaf samples with a high-affinity lysine acetylation enrichment kit.[2] All digests were analyzed using UHPLC–TOF-MS/MS with TIMS. We were very happy with the results:in the mouse liver samples, over 1800 acetylated lysine peptides were identified from 100 μg of starting material over a 50-min gradient, rising to nearly 5000 acetylated lysine peptides using 1000 μg (Figure 3). For the rice leaf samples, 10,000 lysine acetylated sites were identified in a single shot over 4800 protein groups, using a 50-min gradient.
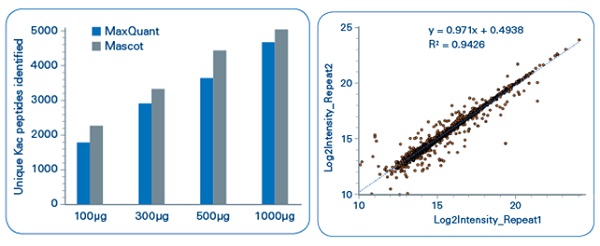
Figure 3: Identification of acetylated lysine peptides enriched from mouse liver, achieved with the help of TIMS. Left: Using four starting amounts. Right: Biological reproducibility.
Conclusion
Directly analyzing proteins from tissue or biofluids can be difficult for a variety of reasons related to genetic and physiological factors—a situation complicated further by the effects of PTMs. HPLC–TOF MS is a powerful technique for uncovering new insights into PTMs, but up until now, its scope has been limited by the capabilities of mass spectrometers.

In this short article, I’ve shown how the combination of HPLC, TIMS, and TOF MS enables proteins or peptides with similar molecular weights to be separated, in situations where previously they would have appeared as the same peak on the mass spectrum. In my view, the ability of the TIMS method to achieve this, while maintaining high speed, sensitivity and reproducibility, means that it has the potential to assist the study of large sample cohorts for clinical research. As a result, it is likely to make a significant contribution to the development of personalized medicine.
References
[1] X. Duet al., Trapped ion mobility spectrometry and PASEF enables in-depth characterization of protein ubiquitination (Poster WP 504), ASMS Conference, 2020, available at www.bruker.com/content/bruker/int/en/landingpages/bdal/poster-hall-asms.html.
[2] X. Liuet al., High sensitivity lysine acetylation profiling with trapped ion mobility spectrometry and PASEF (Poster WP 507), ASMS Conference, 2020, available at www.bruker.com/content/bruker/int/en/landingpages/bdal/poster-hall-asms.html.




{kind=link}