

















Regenerative and cell-based therapies have only recently begun to deliver on the promise they have portended for years, creating opportunities to positively impact patient outcomes and professional success.
In the same way that these new and exciting biological approaches have the potential to revolutionize therapeutics, their very novelty also poses new challenges and opportunities for those of us charged with their development. This is especially true for individuals and teams seeking to move toward the clinic with technology originating from academic collaborations.
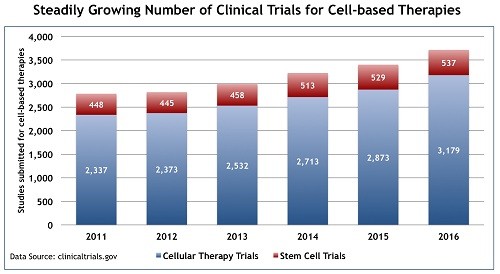 Many of the truly groundbreaking developments in this still-emerging field are originating at a time when many historic funding mechanisms are placing greater demands on entrepreneurship and market-based opportunities for investigator and department alike. The rapid growth and advancement of the technologies, combined with the entrance of new players in this market space, make navigating the regulatory review process quickly and efficiently more pressing than ever.
Many of the truly groundbreaking developments in this still-emerging field are originating at a time when many historic funding mechanisms are placing greater demands on entrepreneurship and market-based opportunities for investigator and department alike. The rapid growth and advancement of the technologies, combined with the entrance of new players in this market space, make navigating the regulatory review process quickly and efficiently more pressing than ever.
If you are a veteran of traditional small molecular pharmaceutical therapies preparing for your first regulatory marathon with the agency, you should familiarize yourself withsome significant and valuable differences between how the process currently plays out in this still-developing area of regulatory review. Here are four tips on how to prepare and use the meeting to your advantage.
1. Take a pre-pre! One of the unique and exciting opportunities available to your translational cell-based program is something only available through the CBER/OCGT offices – literally a pre-, pre-IND meeting to give the agency an opportunity to unofficially review where you are and where you plan to go next, in advance of the formal “Type-B” pre-IND meeting. The meeting is informal, generates no agency minutes, and can be absolutely invaluable in planning your project development milestones well in advance. Prepare for your pre-pre-IND meeting as you would for your pre-IND. In particular, be sure all of your key scientific and regulatory partners are able to participate in the teleconference to make certain you are able to maximize the value of this outstanding opportunity to gain direct guidance and input from the same organization that will ultimately decide the success of your project. A good pre-pre-IND meeting can familiarize you with some of the same reviewers and potential areas of focus you will encounter again at your pre-IND meeting and beyond. Some traditionalists may consider this interaction with the agency to be a luxury. You should view it as a necessity. A few hours of preparation can save you months of work. A few well-considered comments from a reviewer could save you a fortune in development efforts.
2. Get the right team involved at the right time. This is common sense to veterans of CDER review, of course; nonetheless, having a properly built roster of partners carries additional weight in your interactions with the agency when dealing with a field that evolves as rapidly as cell-based and biological therapies. Begin screening for your ideal CMO and preclinical CRO partners early in your project road-map; these groups can be powerful partners at critical phases in the development of your therapies. Your CMO and CRO teams should be able to bring valuable experience when operating within the FDA’s regulatory structure and can help ensure that your developmental milestones are realistic and appropriate. Representatives from your GMP CMO and preclinical CRO should be involved in your preparations for the earliest possible interaction with the agency to help refine your approaches, anticipate issues, and offer justifications to your choices. An experienced regulatory compliance consultant with knowledge of CBER/OCTGT’s evolving approaches to cell-based therapies is always a welcome participant and strengthens your position entering into your interactions with the agency by properly interpreting the written and spoken feedback you will get before, during, and following these meetings. 
3. Manufacturing for the long term. Anyone who has been through the pre-IND process with any branch knows how much emphasis the agency puts on CMC issues, but for your cell-based therapies there is even greater urgency to get an early grip on your process. With many cell-based methods it is almost impossible to avoid animal-origin excipients (including human-derived factors required in many stem cell-based protocols), so of course you will be doing your due diligence in sourcing and compiling all of your CoA and CoO records. But be aware – the agency wants to see xeno-free processes sooner rather than later. Your CMO team may have some experience in possible alternatives worth considering as you build towards your process lock-down. While the agency is not currently demanding xeno-free manufacturing for your cell-based therapies, do not be surprised if they ask whether your team is developing one and how far along your development is. Be prepared with an answer.
4. The rules are different here. In the small-molecule world you come into your pre-IND meeting prepared to discuss your plans to follow the well-worn path the agency has promoted for decades. The numbers, the species, the endpoints – all of these critical study design aspects are largely rote in most cases. With the cell-based therapies things are different. Because of the rapid evolution of the science, the agency is more willing than ever to listen to well-founded scientific proposals and counter-proposals for your critical studies. Make your CRO and CMO collaborators key partners in your process, adding their regulatory and program experience to your scientific expertise. If a single species represents your best in vivo model for both safety and efficacy, you can make that case. If you believe you can best achieve your scientific objectives using only rodents in the preparation of your IND, your preclinical partners can help support your argument in a proposal that can maximize receptivity. If a well-defined agency-recommended release assay is inferior to one you’ve developed in-house, you will get a fair hearing to defend the alternative. The term I have heard over and over again regarding these critical meetings with CBER and OCTGT is “reasonable.” The agency is still in the driver’s seat in reviewing your roadmap to the clinic, but for perhaps the first time, the investigator gets to ride shotgun.
The combination of the rapid increase in our abilities to harness and modify cellular mechanisms and the openness of the commercial market to new entrants create some truly unprecedented opportunities for entrepreneurs and investigators to make an impact with their novel cell-based therapies. As the regulatory environment continues to evolve around these advances, creating the right development program with input from the right support partners can provide a translational pathway for your science that gives you a critical advantage over competitors. Maximize your impact by recognizing how this pathway differs from the traditional IND, and seek to exploit these changes for the benefit of your product, your team, and the ultimate beneficiary – the patient population in need of novel and efficacious technology.



{kind=link}