


















Orphan genetic diseases have become an increasingly popular area for drug development through a variety of novel mechanistictherapeutic approaches.
The development of a means to restore missing proteins to diseased cells consistently and efficiently remains a prominent goal in molecular medicine. While gene therapy has the potential to accomplish this task, considerable obstacles must be overcome before it becomes clinically applicable. Protein therapy on the other hand, comprises the direct delivery of missing proteins to cells circumventing the need for transfer of genetic material into cells, thereby avoiding many of the problems and concerns associated with gene therapy. 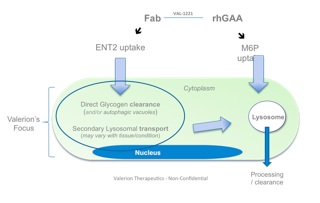
ENT-2 mRNA in human skeletal muscle
Interest in protein therapy has resulted in significant advances in refining protein delivery vehicles. One approach has focused on cell-penetrating peptides, which are short, basic peptides that penetrate living cells. While these peptides are of obvious interest and hold great potential in protein therapy, their use in the treatment of human disease may be limited by documented pro-inflammatory and toxic characteristics.
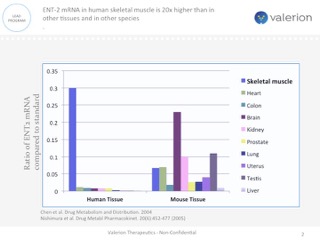
ENT-2 mRNA in human skeletal muscle is 20x higher than in other tissues and in other species
Conversely, employing a novel protein-transduction technology to deliver biologically active proteins to living cells could profoundly impact the clinical viability and efficacy of protein therapy for a wide range of diseases caused by a specific protein deficiency and for diseases where therapies need better uptake into the tissue or organ most likely to benefit from the therapy.
VAL-1221 Fusion Protein Dual sites of action, Complimentary modes of deliver
One approach currently being clinically evaluated is enhanced intracellular delivery of therapeutics via natural transport mechanisms known to be present on cell membranes. Valerion Therapeutics’antibody delivery technology, for example, employs the tissue-localized membrane transporter (ENT2), a receptor which is ubiquitously expressed on cells but naturally enriched in skeletal muscle, liver and central nervous system. The antibody can be genetically fused or chemically conjugated to specific proteins, enzymes and other macromolecules and deliver them to the target tissue, providing a novel way to treat a number of diseases with limited or no current therapeutic options.
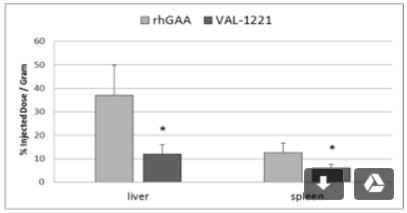
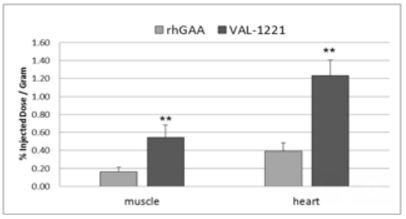
VAL-1221 Compelling Animal Data
Lysosomal storage diseases such as Pompe disease is one example where this approach could add value beyond existing marketed treatments. The development of enzyme replacement therapy has made significant contributions to delaying disease progression by clearing toxic lysosomal glycogen;however, after a period of modest improvement, many patients ultimately progress and resume their downward trajectory in skeletal muscle and respiratory function due to accumulation of extra-lysosomal glycogen.
For sustained clinical benefit, both lysosomal and extra-lysosomal glycogen must be cleared from skeletal muscle. One potential solution is Valerion’s investigational fusion protein, VAL-1221, a therapeutic uniquely designed to target both lysosomal and extra-lysosomal glycogen in skeletal muscle. This is achieved by combining Valerion’s antibody-mediated delivery technology with recombinant human acid alpha-glucosidase (rhGAA) to drive improved delivery of rhGAA to muscle.

Maryze Schoneveld van der Linde, patient living with Pompe Disease
In preclinical studies, VAL-1221 was found to reduce lysosomal glycogen accumulation as effectively as current enzyme replacement therapies, and penetrated living cells independent of the mannose-6-phosphate receptor (M6PR), the mechanism of cell entry employed by current treatments that direct the ERT enzyme to the lysosome. VAL-1221 is currently in clinical studies in patients with late-onset Pompe disease and top-line data are expected in late-2017.
When asked what it’s like to live with Pompe diseaseand her hopes for future therapies, wheelchair-bound Pompe patient MaryzeSchoneveld van der Linde said “Pompeis a very debilitating disease often leading to a loss of limb mobility and breathing function so serious as to require the use of wheelchairs and full time respiratory support, in addition to terrible and constant pain. Pompe dramatically limits quality of life in most patients, and in its most severe forms, can even be fatal. Significant unmet need exists for a variety of new therapeutic options that offer the potential to treat all Pompe patients.”



{kind=link}