
















")
- Kok Li Kwang, West Pharmaceutical Singapore Pte Ltd
- Wee Hoe Tan, West Pharmaceutical Singapore Pte Ltd
- Henry Zhang, West Pharmaceutical Packaging (China) Company Ltd
Based on available data from US FDA between Aug 2017 to Feb 2024, approximately 31% of injectable drug recalls were due to the presence of particulates contamination [1]. Even as improvement measures are being adopted by the pharmaceutical manufacturers to reduce the occurrence of particulate-related recalls, the issue of particulates in drugs continues to be an important topic that is scrutinized by US FDA. In December 2021, US FDA further introduced a draft guidance for industry ‘Inspection of Injectable Products for Visible Particulates’, which emphasizes the importance of using ‘a holistic, risk-based approach to visible particulate control that incorporates product development, manufacturing controls, visual inspection techniques, particulate identification, investigation, and corrective actions designed to assess, correct, and prevent the risk of visible particulate contamination’. Although the United States Pharmacopeial (USP) General Chapters, such as USP <787>, <788> and <789>, have been instrumental in facilitating the particulate control and inspection by drug manufacturers, the new draft guidance also reminds users that meeting the compendial standards are generally not equivalent to meeting the requirements of current Good Manufacturing Practice (cGMP).
According to USP <788>, ‘particulate matter in injections and parenteral infusions consists of extraneous mobile undissolved particles, other than gas bubbles, unintentionally present in the solutions’. The particulates can be classified into three categories:
- Extrinsic, which is foreign to the manufacturing process, such as hair or cellulose.
- Intrinsic, which is present within the manufacturing process or from the packaging materials, such as machine lubricant, packaging glass or rubber.
- Inherent, which is a part of or from the drug formulation, such as protein particles.
Particulates are typically detected during the final inspection of the drug vials prior to product release. In the event that a particulate is found, it should be analyzed to determine its content and nature, which will help in determining the potential source and how the particulate had been introduced or generated. Understanding the source will help in implementing appropriate corrective measures to reduce its recurrence. Extrinsic particulates generally present a greater risk to drug product stability and sterility assurance in comparison to intrinsic and inherent particulates. While it is important to ensure controls over particulate matter contamination, it should also be noted that zero visible particle is not realistic, as there are limitations to current manufacturing and detection technology. In addition, particulate detection is a probabilistic process and particles may be an inherent quality for certain protein therapeutics.
Source and Control Strategy
The presence of unintended particulate matter in parenteral medicine, including injectable drugs, can have severe adverse effects on the patient receiving the drug administration. Particle size can range from nanometers to hundreds of micrometers and may be attributable to various reasons, such as inherent protein aggregate and particles, foreign contaminants, or adsorption of protein molecules to foreign particles [2]. In general, the sources of particulate matter in injectable drugs can be classified into five categories, namely (1) the environment, (2) packaging components, (3) materials and solution used in the manufacture of the drug substance, (4) equipment/machinery and (5) humans. This article examines the contribution of particulate matter from primary packaging components. The primary packaging components coming in direct contact with injectable drugs include elastomeric closures (such as plungers and stoppers) and the container systems (such as vials, syringes, and cartridges).
-
Elastomeric closures
Figure 1 shows the possible sources of particulate matter from elastomeric closures [3]. The six categories include (1) Materials, (2) Equipment, (3) Process, (4) People, (5) Methods and (6) Release-Receipt.
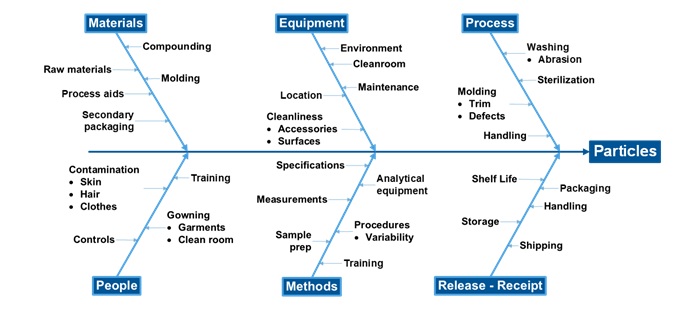
Figure 1 Fishbone diagram showing possible sources of particulate matter from elastomeric closures.
Materials
Elastomeric closures are derived from rubber formulations which consist of various ingredients such as polymer/elastomers, additives, curing agents, accelerators, and antioxidants. Extraneous matter from the raw materials and processing aids could contaminate the elastomeric closures if they are not removed or detected in a timely manner.
The risk to drug product quality is reduced when the primary closure is well understood at the material level using a Quality by Design (QbD) approach. The idea is to link the patient outcome with the product and process. Starting with a clinical goal in mind, Critical Quality Attributes (CQAs) are defined. CQAs are specific properties that must be within a certain range to ensure the desired product quality. For example, a CQA of elastomeric closure might be no more than 3 particulates with size ≥ 25 μm per ml or as otherwise appropriate. The material attributes and process parameters (manufacturing process) that may affect the CQA are then analyzed and appropriate controls are set in place. Low levels of particulates could be achieved through raw material selection, formulation development and process optimization.
Driven primarily by the tighter regulatory expectations, rubber formulations with a more desired performance profile have been developed over the years. An example is the 4040 rubber formulation developed by West Pharmaceutical Services. The QbD approach to development of the 4040 formulation puts focus on mitigating risk from raw material selection to the final product.
Another aspect for consideration is the packaging materials of the raw materials and processing aids. They could also contribute to particulate matter. For example, cellulosic fibers and plastics are common constituent materials used in the packaging of raw materials, i.e. cardboard carton boxes and polybags. If the packaging materials are not handled carefully and segregated procedurally, particulate matter of this nature could contaminate the raw materials as they are being processed downstream.
Equipment
The manufacturing process of packaging components involves various equipment. Cleaning and maintenance of these equipment are critical to avoid contamination and reduce particulate level. Proper line clearance and cleaning should be performed before the manufacture of a different product. Equipment which come into direct contact with products should be inspected regularly for visual cleanliness prior to each use. Unclean surfaces or parts may contribute to particulate matter too. It is recommended under good manufacturing practices (GMP) guidelines to have surfaces in the cleanroom that are designed to be easy to clean and sanitize, and does not allow the build-up of contamination [4].
The machinery involved in the production of the elastomeric closures should be regularly inspected and maintained to ensure that they are operating under optimal conditions. Mechanical deterioration in the form of fatigue, wear and tear, abrasion, and corrosion of the equipment parts over time can result in particle generation due to the surface breakdown within the machine, also known as wear debris [5].
The cleanroom environment which houses the equipment and machinery plays an important role in controlling particulate matter load. According to ISO 14644-1:2015, the definition of a cleanroom is as follows, “room within which the number concentration of airborne particles is controlled and classified, and which is designed, constructed and operated in a manner to control the introduction, generation and retention of particles inside the room” [6]. Cleanrooms typically require a certain airflow design, known as the cascading pressure. This means that the cleanroom with critical processes such as aseptic fill-finish and sterile material addition has the highest level of pressure and the cleanroom with the least critical operations comparatively has the lowest level of pressure. This ensures a positive airflow pressure is always flowing from the most stringent cleanroom to the less stringent cleanroom, preventing any flow of contamination into the cleanroom with the critical operations. For example, elastomeric closures may be loaded into the pharmaceutical washer from an ISO 8 cleanroom and unloaded and packed in an ISO 5 cleanroom.
The environment and air supply system should be monitored and controlled. Monitoring data, such as real-time/continuous total particle counts can be used to facilitate appropriate responses, which typically include a root cause investigation into the deviation from the control state and possible impact assessment. Specified technical control measures should form the basis of process design to deliver a state of product manufacture under environmentally controlled conditions with a low (reduced or eliminated) risk of contamination [7].
Process
The typical manufacturing process of an elastomeric closure involves multiple steps including mixing, calendaring, molding, trimming, washing, packaging, and sterilization. There are several points in the process which particulate matter can potentially contaminate the rubber components. During the mixing stage where the ingredients are mixed together, there is potential risk that foreign particulates such as hair, fibers may get into the formulation. The formulation should also be well mixed to ensure a homogeneous dispersion. At the molding step, the rubber sheets from the calendaring step are loaded onto the molds and are subjected to heat and pressure to turn the rubber into the desired shape. Vulcanization takes place where chemical cross-links are formed to give the formulation its thermoset elastomeric properties. Foreign particulates introduced at this stage could be embedded in the rubber components. Contamination control strategy should be used to avoid the introduction of particulates during these processing steps.
Next, at the trimming stage, the technicians visually inspect the molded sheets and cut out any individual components that have visual defects such as partially-formed stoppers. Then, the molded sheets are placed on the trimming system and the rubber components are trimmed from the web. This is a critical step because improper trimming may result in defective components such as trim fringes and may increase the particle load for the produced batch.
Depending on the intended application requirement, the rubber components may be coated with silicone oil. This is typically applied before or during the washing stage (by tumbling or emulsification). Silicone oil increases the lubricity properties of the rubber components but poses a concern with silicone oil droplets which contribute to the population of sub-visible particles in finished drug products. Silicone oil is known to have undesired interactions with protein-based drugs, affecting the chemical and physical stability of the drug formulation [8]. Fortunately, the pharmaceutical packaging industry has successfully developed processes to optimize the siliconization process and minimize the generation of silicone oil droplets. For example, the use of cross-linkable polydimethylsiloxane coating is developed to address the concern of silicone oil droplets by the application of UV rays and heat after spray coating on the molded panels. This curing process improves the coating-to-elastomer adherence and greatly reduces potential for release of subvisible particles as compared to conventional silicone oil [9].
The use of fluoropolymer film such as ethylene tetrafluoroethylene (ETFE) on elastomeric closure surface is another way to mitigate the particulate matter. The film provides an effective barrier against organic and inorganic extractables. Additionally, because of very low surface energy, it minimizes interaction between the drug and the closure and reduces the risk of particulate formation. Interaction was evaluated by measurement of particulates, turbidity, and recovery of drug products (simulated/commercial) under agitated/stressed conditions. For a variety of proteins, stoppers with fluoropolymer film resulted in lower levels of particulate formation, lower turbidity, and higher protein recovery. These results demonstrate reduced interaction with drug product and risk of particulate from elastomers [10].
Ready-to-sterilize and ready-to-use rubber components undergo a washing process after trimming. This is an important step in controlling the particle load of the manufactured batch. If the washing process is not optimized, it could result in a higher particle load. Particulate matter may be generated during the washing process as the rubber components rub against one another or the stainless steel surface of equipment as they are being tumbled around during the wash cycle and during the transfer in and out of the washer. Different manufacturers and facilities may differ in the washers used and washing cycles. Regardless of the equipment and washing procedures, it is vital to validate the washing process and ensure that the washed rubber components meet the specifications for bioburden, endotoxin, particles and where applicable, silicone oil level.
Another approach to significantly minimize the presence of particles and defects is to use automatic, program-controlled vision inspection systems to inspect all surfaces of elastomeric components in an ISO 5 clean room environment after washing. This process enhances the quality of packaging components by removing components with adhered and embedded particulate matter, molding and/or trimming defects prior to packaging. It assists in reducing the total cost of goods by minimizing the risk of rejecting drug products due to visible particulates and closure defects [11].
People
Even though manufacturing technology has advanced immensely over the years and the implementation of automation robotics has reduced the level of human activity and manual intervention, people are still very much needed in the manufacturing of elastomeric closures. For example, humans are needed to operate and maintain the equipment and machinery, monitor and ensure that the automated processes are running smoothly, perform manual processes such as weighing and adding of the raw materials in the right sequence and amount, visual inspection of the molded sheets and cutting any defective closures.
The presence of human personnel in the cleanroom is usually the main contributor of airborne particulates and microbial contamination. An average human sheds about 200, 000, 000 skin cells every hour [12]. The human body continuously undergoes regenerative processes to produce skin flakes, oil, sweat and hair [13]. There are other sources of contamination associated with humans. The clothing that we wear are a source of fiber strands.
Most airborne particulates will settle due to gravity and the settling rate is dependent on the particle size [14]. However, human activity and motion in the cleanroom may stir up any settled airborne particulates due to the turbulent air motion created [15]. When humans are walking briskly (5 miles/hour), about 7.5 million particles are shed as compared to 5 million particles when walking slowly (2 miles/hour) [16]. Therefore, good personnel practices and stringent procedures are required to reduce the particulate risk.
Special clothing designed for the clean environments is required to prevent the particulate contamination from human body [17]. To be effective, the clothing must form a particulate barrier for the human micro-environment and avoid being a significant particulate contributor itself. Selecting a suitable material for a gown is important to minimize particle shedding. The frequency of change of reusable gowns, cleaning, processing, packing of gowns should be defined, and sufficient training should be in place to ensure correct gowning procedure is followed consistently.
-
Glass container
Apart from elastomeric closures, the consideration of particulate matter is not complete without examining the drug product and its container system in its entirety.
Biotherapeutic drugs have an inherent characteristic to aggregate and form protein particles. The aggregation can occur at any stage in the drug manufacturing process including fermentation, purification, formulation and storage. External factors such as heating, freeze-thaw, mechanical stress, ultraviolet light, and exposure to extractables and leachables are examples of external factors that can result in protein aggregation [18-19]. In a study conducted by Krayukhina et. Al., the adsorption of protein to the unlubricated glass surface could be attributed to charge-charge effects whereas for unlubricated polymer surface, protein could be adsorbed to the surface mainly via hydrophobic interactions. It was also suggested that the presence of air bubbles is a prerequisite for the formation of protein particles [20]. Protein molecules tend to adsorb onto interfaces such as air-liquid, container surface-liquid and liquid-liquid in vials and syringes. Once adsorbed, the protein molecules denature, undergo conformational change and aggregate as protein particles which are detected as sub-visible particles [19]. Particle counts from silicone-oil lubricated syringes were higher than those silicone-oil free syringes. This is because silicone oil lubrication increases the adsorption of protein to the surface and/or disrupts the formed protein layer subsequently [20].
Glass has been used as a packaging material for pharmaceutical drugs and biologic formulations due to its inherent properties. It is non-porous and impermeable to gases, transparent, chemically resistant to organics, dimensionally stable and has high tensile strength. However, despite of the advantages, there are challenges in using glass as a packaging material for injectable drugs, with regards to the topic of particulate matter.
One source of glass particulate matter is delamination from glass containers. Delamination refers to the flaking of the upper surface of the glass from the glass container. This is often observed as glass flakes (also known as lamellae) in the solution. It is often caused by a combination of factors including glass material composition and chemistry, formation and treatment of the glass container and reaction of the glass with the drug product.
In terms of glass material composition and chemistry, soda-lime-silica glass has lower hydrolytic resistance as compared to borosilicate glass. Ammonium sulfate is added prior to the glass annealing process during the vial manufacturing process to react with sodium ions near the glass surface to produce sodium sulfate. Sodium sulfate is highly soluble in water and is washed away during the subsequent washing step. This results in surface pitting for ammonium sulfate-treated vials which have a higher tendency to undergo delamination [21].
For more than the past decade, USP General Chapter <660> has categorized glass material used for pharmaceutical products into 3 types, namely Type I glass which is borosilicate glass, Type II glass which is treated soda-lime silica glass and Type III glass which is soda-lime silica glass. In the past several years, new developments in the glass sector have seen new glass composition. One example is the Corning® Valor® Glass Vials from Corning Incorporated. Corning® Valor® Glass Vials represent a paradigm shift in pharmaceutical glass packaging, utilizing cutting-edge glass science to enhance the preservation and delivery of drugs. The chemical composition of Corning® Valor® Glass Vial is aluminosilicate. Figure 2 shows the chemical structure of borosilicate glass, aluminosilicate glass and soda-lime glass. The Boron atoms in borosilicate glass have been replaced by the Aluminum atoms in aluminosilicate glass [22]. One reason for glass delamination in borosilicate glass is the formation of heterogenous surface chemistry on the vial during the converting process. Boron-containing compounds adhere to the walls and heel of the vial which increases the risk to glass delamination as seen in Figure 3 [23]. Aluminum replaces boron in aluminosilicate glass which ensures that the glass surface chemistry is uniform after the conversion process.
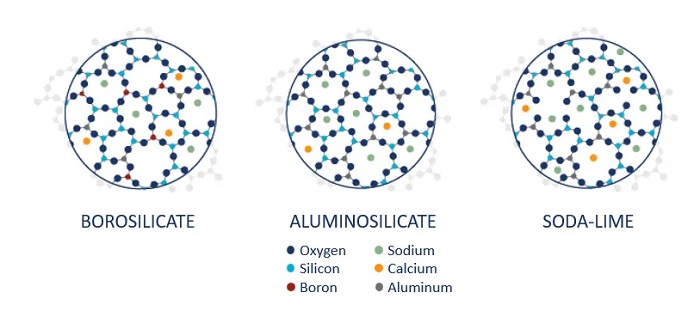
Figure 2 Types of glass composition.

Figure 3 Comparison of Borosilicate glass and Corning® Valor® glass. During the borosilicate glass conversion process, volatilization of the boron element occurs which is evident by the formation of blue vapor. This results in a heterogeneous inner surface chemistry on the vial. This is not observed in the conversion process for aluminosilicate glass as the Boron elements have been replaced by Aluminum elements.
USP General Chapter <660> was revised to remove glass classifications based on composition in Oct 2023. Under the proposed compendial revisions, USP General Chapter <660> would instead define glass Types I, II, and III by performance characteristics, allowing for additional compositions to be considered Type I, II, and III glass. There are no proposed changes to test procedures or acceptance criteria [24]. In addition, FDA is supportive of the use of new glass compositions if they demonstrate equivalent or superior performance characteristics such as improved thermal and hydrolytic resistance compared to the current compendial glass compositions and demonstrate suitability for the drug product [25].
Glass is formed by two primary methods – tubular glass and molded glass. The annealing process to mold and shape the glass mold or glass tube into the desired dimension and design involves high temperatures (500°C-600°C). The heating of the glass during the forming process facilitates the migration of alkali borates from the silica network to the glass surface, causing a localized enrichment [20]. The heel regions are particularly enriched in sodium and boron and have lower chemical durability and undergo flaking at higher rates than the surrounding glass due to the higher temperature required to form the vial bottom [20, 26]. Surface treatment including siliconization and lamination coating contribute to the glass delamination too. The treatment alters the glass surface chemistry and accelerates the corrosion rates when the altered glass surface coming into contact with the drug formulation [26].
It is common for soda-lime glass to undergo reaction with the drug products due to the lower hydrolytic resistance. This often forms a layer of silica-rich layer that eventually detaches from the glass bulk. Leachables from the packaging components (such as elastomeric closures, glass or plastic containers) can react with the drug product and form precipitates on the glass wall [26]. Other factors which could affect delamination include, concentration, pH and ionic strength of the drug formulation, sterilization process and storage conditions [27].
Delamination is an extremely serious issue that can result in drug recalls. Proteins may preferentially adsorb to glass more as compared to polymer systems due to the difference in surface characteristics and induce subvisible aggregates. New cyclic olefin polymer (COP) resin offerings may avoid the delamination issue and provide reduced protein aggregation of drug product due to the unique surface properties of the COP material. Work has demonstrated that for several therapeutic proteins, a container system comprising COP vials and fluoropolymer film laminated stoppers results in a far lower level of particle formation under stress, than a comparable glass system [28].
Another area in the drug manufacturing process that could produce significant particulate matter is the glass-to-glass vial contact along the filling lines. Vial-to-vial interactions along the filling line are classified as frictive sliding or impact events. Frictive sliding between the glass vials can result in fracture and produce extensive particles in the subvisible range (< 50 µm). Impact events are used to describe higher tensile stresses on the glass surface that could result in the propagation of single microscopic flaw or convergence of multiple microscopic flaws. This usually results in the forming of glass chips and visible particles (> 100 µm) [29]. There are known and effective mitigations for particulate generation due to glass-to-glass contact. A layer of coating can be applied on the external surface of the vials. This has been proven to lower the coefficient of friction and minimize the generation of particulates when the vials contact each other. The packaging configuration of the vials can be optimized by using nested configurations such as nest and tub or cup nest to eliminate glass-to-glass contact. This reduces risk of glass breakages and cosmetic defects.
Conclusion
Many factors throughout a drug’s manufacturing and storage may impact the level of particulate in the final drug product. Potential particulate contamination from the direct contact materials, such as the fill-finish equipment and packaging components such as closures and containers, as well as indirect contact attributable from upstream materials and processes may pose a risk to the final drug product and patient safety. While the concept of zero particulates is idealistic at this juncture due to the constraints from technology and resource availability, it is important to understand the risks from the various factors and deploy appropriate controls to mitigate these risks. These can be in the form of additional visual inspection at relevant process steps, utilizing fit-for-purpose materials which are optimized to reduce particulate generation, improving process parameters and equipment design to meet critical quality requirements and many more. The ultimate goal is to continuously pursue improvements in order to achieve successful treatment outcome and patient safety.
References
- https://www.fda.gov/drugs/drug-safety-and-availability/drug-recalls and https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/recalls-biologics (Accessed February 02, 2024, data available from August 30, 2017 to February 02, 2024)
- John Rech, Amber Fradkin, Aaron Krueger, Crystal Kraft & Diane Paskiet. (2020). Evaluation of Particle Techniques for the Characterization of Subvisible Particles From Elastomeric Closure Components. Journal of Pharmaceutical Sciences VOLUME 109, ISSUE 5, P1725-1735, MAY 01, 2020. https://doi.org/10.1016/j.xphs.2020.01.026
- Unique Considerations for Testing and Understanding Particle Loads for Primary Packaging Components. AAPS Workshop on Challenges and Patient Impact of Particles in Biologics. April 30, 2017
- John Hamlin. (2004). Overview of Aseptic Fill/Finish Manufacturing. BioRealty Inc. https://www.biorealty.com/blog/overview-of-aseptic-fillfinish-manufacturing
- Bennett Fitch. (2013). Anatomy of Wear Debris. https://www.machinerylubrication.com/Read/29537/wear-debris-anatomy
- ISO 14644-1:2015. (2015). Cleanrooms and associated controlled environments — Part 1: Classification of air cleanliness by particle concentration. https://www.iso.org/obp/ui/#iso:std:iso:14644:-1:ed-2:v1:en
- James L. Drinkwater. The challenges of risk based environmental monitoring in sterile product filling. European Pharmaceutical Review, 29 February 2016.
- Yoshino et al. (2014). Functional Evaluation and Characterization of a Newly Developed Silicone Oil-Free Prefillable Syringe System. Journal of Pharmaceutical Sciences VOLUME 103:1520–1528, 2014. https://doi.org/10.1002/jps.23945
- Technical Report 2000/026 TR 2000026 B2-Coating Quantitative Particle Analysis. West Pharmaceutical Services, Inc.
- Ranjana Singh, William Garzon-Rodriguez, etc. Elastomer Stoppers with FluroTec® Film: The Right Choice for SARS-CoV-2 Vaccines. West Pharmaceutical Services, Inc.
- Technical Report 2010/137 Utilizing West Envision™ as a Risk Mitigation Solution. West Pharmaceutical Services, Inc.
- Imperial College London. (2016, November 29). New insights into skin cells could explain why our skin doesn’t leak. ScienceDaily. Retrieved June 22, 2022. www.sciencedaily.com/releases/2016/11/161129114910.htm
- Damon Larkin. (n.d.). The contamination risks posed by laundered cleanroom apparel. https://beta-static.fishersci.com/content/dam/fishersci/en_US/documents/programs/scientific/technical-documents/white-papers/kimberly-clark-contamination-risks-white-paper.pdf
- Trevor. (2021, October 05). Basic Clean Room Requirements, Designs for GMP Clean Rooms. PharmOut. https://www.pharmout.net/basic-cleanroom-requirements
- Terra Universal. (n.d.) 7 Strategies to Better Cleanroom Gowning. https://www.terrauniversal.com/blog/7-strategies-to-better-gowning
- Andrew Ramage. (n.d.) The Weakest Link In The Cleanroom Environment? Humans. Cherwell Laboratories. https://www.cherwell-labs.co.uk/cherwell-labs-post/weakest-link-in-cleanroom-environment-humans
- Release of sterile medicinal products – looking at the focal points. Tim Sandle, European Pharmaceutical Review, 25 August 2020.
- Joubert, M. K., Luo, Q., Nashed-Samuel, Y., Wypych, J., & Narhi, L. O. (2011). Classification and characterization of therapeutic antibody aggregates. The Journal of biological chemistry, 286(28), 25118–25133. https://doi.org/10.1074/jbc.M110.160457
- Kiyoshi, M., Shibata, H., Harazono, A., Torisu, T., Maruno, T., Akimaru, M., Asano, Y., Hirokawa, M., Ikemoto, K., Itakura, Y., Iwura, T., Kikitsu, A., Kumagai, T., Mori, N., Murase, H., Nishimura, H., Oda, A., Ogawa, T., Ojima, T., Okabe, S., … Ishii-Watabe, A. (2019). Collaborative Study for Analysis of Subvisible Particles Using Flow Imaging and Light Obscuration: Experiences in Japanese Biopharmaceutical Consortium. Journal of pharmaceutical sciences, 108(2), 832–841. https://doi.org/10.1016/j.xphs.2018.08.006
- Krayukhina, E., Tsumoto, K., Uchiyama, S., & Fukui, K. (2015). Effects of syringe material and silicone oil lubrication on the stability of pharmaceutical proteins. Journal of pharmaceutical sciences, 104(2), 527–535. https://doi.org/10.1002/jps.24184
- Zhao, J., Lavalley, V., Mangiagalli, P., Wright, J. M., & Bankston, T. E. (2014). Glass delamination: a comparison of the inner surface performance of vials and pre-filled syringes. AAPS PharmSciTech, 15(6), 1398–1409. https://doi.org/10.1208/s12249-014-0167-y
- Dr. Bettine Boltres (2023, October 02). USP<660> – Time for a Change. www.westpharma.com. https://www.westpharma.com/blog/2023/october/usp-revision-glass-container-vials-packaging-chapter-660
- Corning® Valor® Glass Delamination Report Customer Information Letter, CPT_BF_Delamination_13July2018
- USP <660> Containers-Glass, United States Pharmacopeia (2023), Current DocID: GUID-67466D06-7DCC-46C2-A61F-013D827120C6_2_en-US
- Ashley B. Boam (2022) Ashley B. Boam to Ms. Jessica Simpson, 01 November 2022 [Letter] U.S. Food & Drug Administration, Ref: 11-22-002-AB. Available from: https://www.uspnf.com/sites/default/files/usp_pdf/EN/notices/2023/glass_REF11-22-002-AB.pdf [Accessed 20 May 2024]
- Schaut, R. A., & Weeks, W. P. (2017). Historical Review of Glasses Used for Parenteral Packaging. PDA journal of pharmaceutical science and technology, 71(4), 279–296. https://doi.org/10.5731/pdajpst.2016.007377
- Daniele Z. (n.d.) Glass Delamination: Risks, Reality and Regulatories [PowerPoint Slides] Nuova OMPI – Stevanato Group. https://www.pda.org/docs/default-source/website-document-library/chapters/presentations/metro/glass-delamination-risks-reality-and-regulatories.pdf?sfvrsn=2dc5a38e_6
- Ranjana Singh, Cathy Zhao and Lloyd Waxman Fran L. Effect of Container Surface on Protein Aggregation. West Pharmaceutical Services, Inc.
- Timmons, C. L., Liu, C. Y., & Merkle, S. (2017). Particulate Generation Mechanisms during Bulk Filling and Mitigation via New Glass Vial. PDA journal of pharmaceutical science and technology, 71(5), 379–392. https://doi.org/10.5731/pdajpst.2017.007724


{kind=link}